David E. Mortenson, Gabriel J. Brighty, Lars Plate, Grant Bare, Wentao Chen, Suhua Li, Hua Wang, Benjamin F. Cravatt, Stefano Forli , Evan T. Powers, K. Barry Sharpless Ian A. Wilson, and Jeffery W. Kelly
J. Am. Chem. Soc., Article ASAP
DOI: 10.1021/jacs.7b08366
Drug candidates are generally discovered using biochemical screens employing an isolated target protein or by utilizing cell-based phenotypic assays. Both noncovalent and covalent hits emerge from such endeavors. Herein, we exemplify an “Inverse Drug Discovery” strategy in which organic compounds of intermediate complexity harboring weak, but activatable, electrophiles are matched with the protein(s) they react with in cells or cell lysate. An alkyne substructure in each candidate small molecule enables affinity chromatography–mass spectrometry, which produces a list of proteins that each distinct compound reacts with. A notable feature of this approach is that it is agnostic with respect to the cellular proteins targeted. To illustrate this strategy, we employed aryl fluorosulfates, an underexplored class of sulfur(VI) halides, that are generally unreactive unless activated by protein binding. Reversible aryl fluorosulfate binding, correct juxtaposition of protein side chain functional groups, and transition-state stabilization of the S(VI) exchange reaction all seem to be critical for conjugate formation. The aryl fluorosulfates studied thus far exhibit chemoselective reactivity toward Lys and, particularly, Tyr side chains, and can be used to target nonenzymes (e.g., a hormone carrier or a small-molecule carrier protein) as well as enzymes. The “Inverse Drug Discovery” strategy should be particularly attractive as a means to explore latent electrophiles not typically used in medicinal chemistry efforts, until one reacts with a protein target of exceptional interest. Structure–activity data can then be used to enhance the selectivity of conjugate formation or the covalent probe can be used as a competitor to develop noncovalent drug candidates. Here we use the “Inverse Drug Discovery” platform to identify and validate covalent ligands for 11 different human proteins. In the case of one of these proteins, we have identified and validated a small-molecule probe for the first time.
Sunday, December 31, 2017
Saturday, December 16, 2017
Selectively targeting the kinome-conserved lysine of PI3Kδ as a general approach to covalent kinase inhibition
Samuel E Dalton, Lars Dittus, Daniel A. Thomas, Maire A. Convery, Joao Nunes, Jacob T. Bush, John P. Evans, Thilo Werner, Marcus Bantscheff, John A. Murphy, and Sebastien Campos
J. Am. Chem. Soc., 2017
DOI: 10.1021/jacs.7b08979
Selective covalent inhibition of kinases by targeting poorly conserved cysteines has proven highly fruitful to date in the development of chemical probes and approved drugs. However, this approach is limited to ~200 kinases possessing such a cysteine near the ATP-binding pocket. Herein, we report a novel approach to achieve selective, irreversible kinase inhibition, by targeting the conserved catalytic lysine residue. We have illustrated our approach by developing selective, covalent PI3Kδ inhibitors that exhibit nanomolar potency in cellular assays, and a duration of action >48 h in CD4+ T cells. Despite conservation of the lysine residue throughout the kinome, the lead compound shows high levels of selectivity over a selection of lipid and protein kinases in biochemical assays, as well as covalent binding to very few off-target proteins in live-cell proteomic studies. We anticipate this approach could offer an alternative general strategy, to targeting non-conserved cysteines, for the development of selective covalent kinase inhibitors.
J. Am. Chem. Soc., 2017
DOI: 10.1021/jacs.7b08979
Selective covalent inhibition of kinases by targeting poorly conserved cysteines has proven highly fruitful to date in the development of chemical probes and approved drugs. However, this approach is limited to ~200 kinases possessing such a cysteine near the ATP-binding pocket. Herein, we report a novel approach to achieve selective, irreversible kinase inhibition, by targeting the conserved catalytic lysine residue. We have illustrated our approach by developing selective, covalent PI3Kδ inhibitors that exhibit nanomolar potency in cellular assays, and a duration of action >48 h in CD4+ T cells. Despite conservation of the lysine residue throughout the kinome, the lead compound shows high levels of selectivity over a selection of lipid and protein kinases in biochemical assays, as well as covalent binding to very few off-target proteins in live-cell proteomic studies. We anticipate this approach could offer an alternative general strategy, to targeting non-conserved cysteines, for the development of selective covalent kinase inhibitors.
Wednesday, December 6, 2017
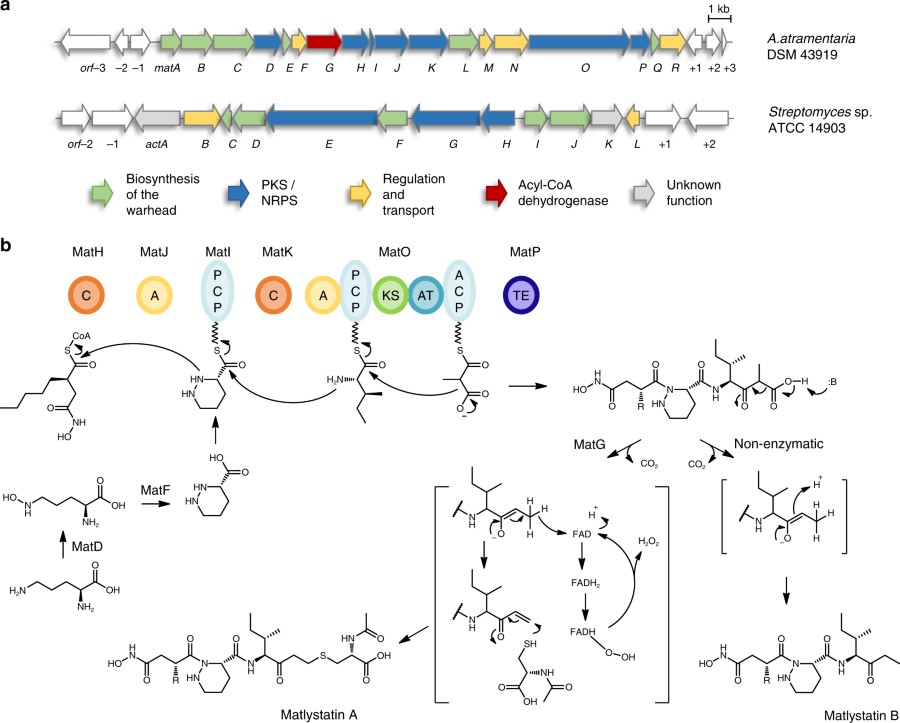
Warhead biosynthesis and the origin of structural diversity in hydroxamate metalloproteinase inhibitors
Franziska Leipoldt, Javier Santos-Aberturas, Dennis P. Stegmann, Felix Wolf, Andreas Kulik, Rodney Lacret, Désirée Popadić, Daniela Keinhörster, Norbert Kirchner, Paulina Bekiesch, Harald Gross, Andrew W. Truman & Leonard Kaysser
doi: 10.1038/s41467-017-01975-6
Metalloproteinase inhibitors often feature hydroxamate moieties to facilitate the chelation of metal ions in the catalytic center of target enzymes. Actinonin and matlystatins are potent metalloproteinase inhibitors that comprise rare N-hydroxy-2-pentyl-succinamic acid warheads. Here we report the identification and characterization of their biosynthetic pathways. By gene cluster comparison and a combination of precursor feeding studies, heterologous pathway expression and gene deletion experiments we are able to show that the N-hydroxy-alkyl-succinamic acid warhead is generated by an unprecedented variation of the ethylmalonyl-CoA pathway. Moreover, we present evidence that the remarkable structural diversity of matlystatin congeners originates from the activity of a decarboxylase-dehydrogenase enzyme with high similarity to enzymes that form epoxyketones. We further exploit this mechanism to direct the biosynthesis of non-natural matlystatin derivatives. Our work paves the way for follow-up studies on these fascinating pathways and allows the identification of new protease inhibitors by genome mining.
Tuesday, December 5, 2017
Chemoselective Installation of Amine Bonds on Proteins through Aza-Michael Ligation
Allyson M. Freedy, Maria J. Matos, Omar Boutureira, Francisco Corzana, Ana Guerreiro, Padma Akkapeddi, Víctor J. Somovilla, Tiago Rodrigues, Karl Nicholls, Bangwen Xie, Gonzalo Jiménez-Osés, Kevin M. Brindle, André A. Neves, & Gonçalo J. L. Bernardes
J. Am. Chem. Soc., 2017 ASAP
DOI: 10.1021/jacs.7b10702
Chemical modification of proteins is essential for a variety of important diagnostic and therapeutic applications. Many strategies developed to date lack chemo- and regioselectivity as well as result in non-native linkages that may suffer from instability in vivo and adversely affect the protein’s structure and function. We describe here the reaction of N-nucleophiles with the amino acid dehydroalanine (Dha) in a protein context. When Dha is chemically installed in proteins, the addition of a wide-range N-nucleophiles enables the rapid formation of amine linkages (secondary and tertiary) in a chemoselective manner under mild, biocompatible conditions. These new linkages are stable at a wide range of pH values (pH 2.8 to 12.8), under reducing conditions (biological thiols such as glutathione) and in human plasma. This method is demonstrated for three proteins and is shown to be fully compatible with disulfide bridges, as evidenced by the selective modification of recombinant albumin that displays 17 structurally relevant disulfides. The practicability and utility of our approach is further demonstrated by the construction of a chemically modified C2A domain of Synaptotagmin-I protein that retains its ability to preferentially bind to apoptotic cells at a level comparable to the native protein. Importantly, the method was useful for building a homogeneous antibody-drug conjugate with a precise drug-to-antibody ratio of 2. The kinase inhibitor crizotinib was directly conjugated to Dha through its piperidine motif, and its antibody-mediated intracellular delivery results in 10-fold improvement of its cancer cell-killing efficacy. The simplicity and exquisite site-selectivity of the aza-Michael ligation described herein allows the construction of stable secondary and tertiary amine-linked protein conjugates without affecting the structure and function of biologically relevant proteins.

Subscribe to:
Posts (Atom)
Linking of fragments in neighboring binding sites is one of the optimization strategies in fragment-based drug discovery, where additive or even more substantial bioactivity improvements can be realized. However, such efforts present a considerable challenge when one fragment binds covalently to the target protein, as small modifications can influence the correct positioning of the covalent warhead toward the targeted nucleophilic residue. Here, we present a case study of fragment linking that yielded single-digit micromolar, covalent inhibitors of the SARS-CoV-2 main protease, starting from fragments that were inactive in the biochemical assay. Using structural information from a recent, high-throughput crystallographic fragment screen, we show that the success of fragment linking in the design of targeted covalent inhibitors is heavily impacted by several factors, including the warhead type, the labeling chemistry, and even subtle changes in the designed linker. Notably, we observe that induced fit effects might override the original fragment orientations in the linked molecule, highlighting the need for reliable structure verification, especially in consecutive rounds of fragment elaboration.
Levente Kollár, Levente M. Mihalovits, Dávid Bajusz, DamijanKnez, József Simon, Blake H. Balcomb, Daren Fearon, Stanislav Gobec, György M. K...

-
Design, synthesis and biological evaluation of the activity-based probes for FGFR covalent inhibitorDandan Zhu, Zijian Zheng, Huixin Huang, Xiaojuan Chen, Shuhong Zhang, Zhuchu Chen, Ting Liu, Guangyu Xu, Ying Fu, Yongheng Chen, European Jo...
-
Yoav Shamir, Nir London bioRxiv 2025.03.19.642201 doi: https://doi.org/10.1101/2025.03.19.642201 Recent years have seen an explosion in the...
-
DOI Ansgar Oberheide, Maxime van den Oetelaar, Jakob Scheele, Jan Borggräfe, Semmy Engelen, Michael Sattler, Christian Ottmann, ...
