José L. Montaño, Brian J. Wang, Regan F. Volk, Sara E. Warrington, Virginia G. Garda, Katherine L. Hofmann, Leo C. Chen, and Balyn W. Zaro
ACS Chemical Biology 2022
DOI: 10.1021/acschembio.1c00980
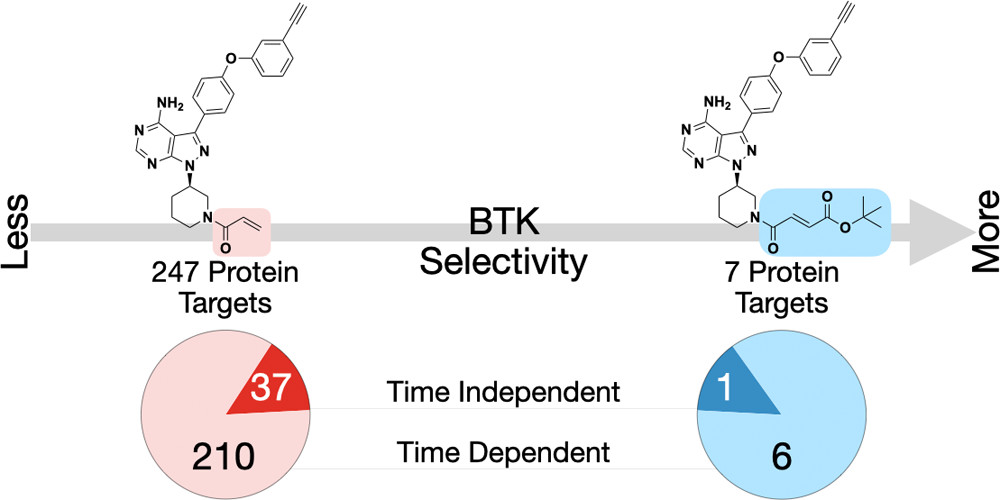
Covalent inhibitors are viable therapeutics. However, off-target reactivity challenges the field. Chemists have attempted to solve this issue by varying the reactivity attributes of electrophilic warheads. Here, we report the development of an approach to increase the selectivity of covalent molecules that is independent of warhead reactivity features and can be used in concert with existing methods. Using the scaffold of the Bruton’s tyrosine kinase (BTK) inhibitor Ibrutinib for our proof-of-concept, we reasoned that increasing the steric bulk of fumarate-based electrophiles on Ibrutinib should improve selectivity via the steric exclusion of off-targets but retain rates of cysteine reactivity comparable to that of an acrylamide. Using chemical proteomic techniques, we demonstrate that elaboration of the electrophile to a tert-butyl (t-Bu) fumarate ester decreases time-dependent off-target reactivity and abolishes time-independent off-target reactivity. While an alkyne-bearing probe analogue of Ibrutinib has 247 protein targets, our t-Bu fumarate probe analogue has only 7. Of these 7 targets, BTK is the only time-independent target. The t-Bu inhibitor itself is also more selective for BTK, reducing off-targets by 70%. We investigated the consequences of treatment with Ibrutinib and our t-Bu analogue and discovered that only 8 proteins are downregulated in response to treatment with the t-Bu analogue compared to 107 with Ibrutinib. Of these 8 proteins, 7 are also downregulated by Ibrutinib and a majority of these targets are associated with BTK biology. Taken together, these findings reveal an opportunity to increase cysteine-reactive covalent inhibitor selectivity through electrophilic structure optimization.