Christian Dubiella, Benika J. Pinch, Kazuhiro Koikawa, Daniel Zaidman, Evon Poon, Theresa D. Manz, Behnam Nabet, Shuning He, Efrat Resnick, Adi Rogel, Ellen M. Langer, Colin J. Daniel, Hyuk-Soo Seo, Ying Chen, Guillaume Adelmant, Shabnam Sharifzadeh, Scott B. Ficarro, Yann Jamin, Barbara Martins da Costa, Mark W. Zimmerman, Xiaolan Lian, Shin Kibe, Shingo Kozono, Zainab M. Doctor, Christopher M. Browne, Annan Yang, Liat Stoler-Barak, Richa B. Shah, Nicholas E. Vangos, Ezekiel A. Geffken, Roni Oren, Eriko Koide, Samuel Sidi, Ziv Shulman, Chu Wang, Jarrod A. Marto, Sirano Dhe-Paganon, Thomas Look, Xiao Zhen Zhou, Kun Ping Lu, Rosalie C. Sears, Louis Chesler, Nathanael S. Gray & Nir London
Nat Chem Biol. 2021
DOI https://doi.org/10.1038/s41589-021-00786-7
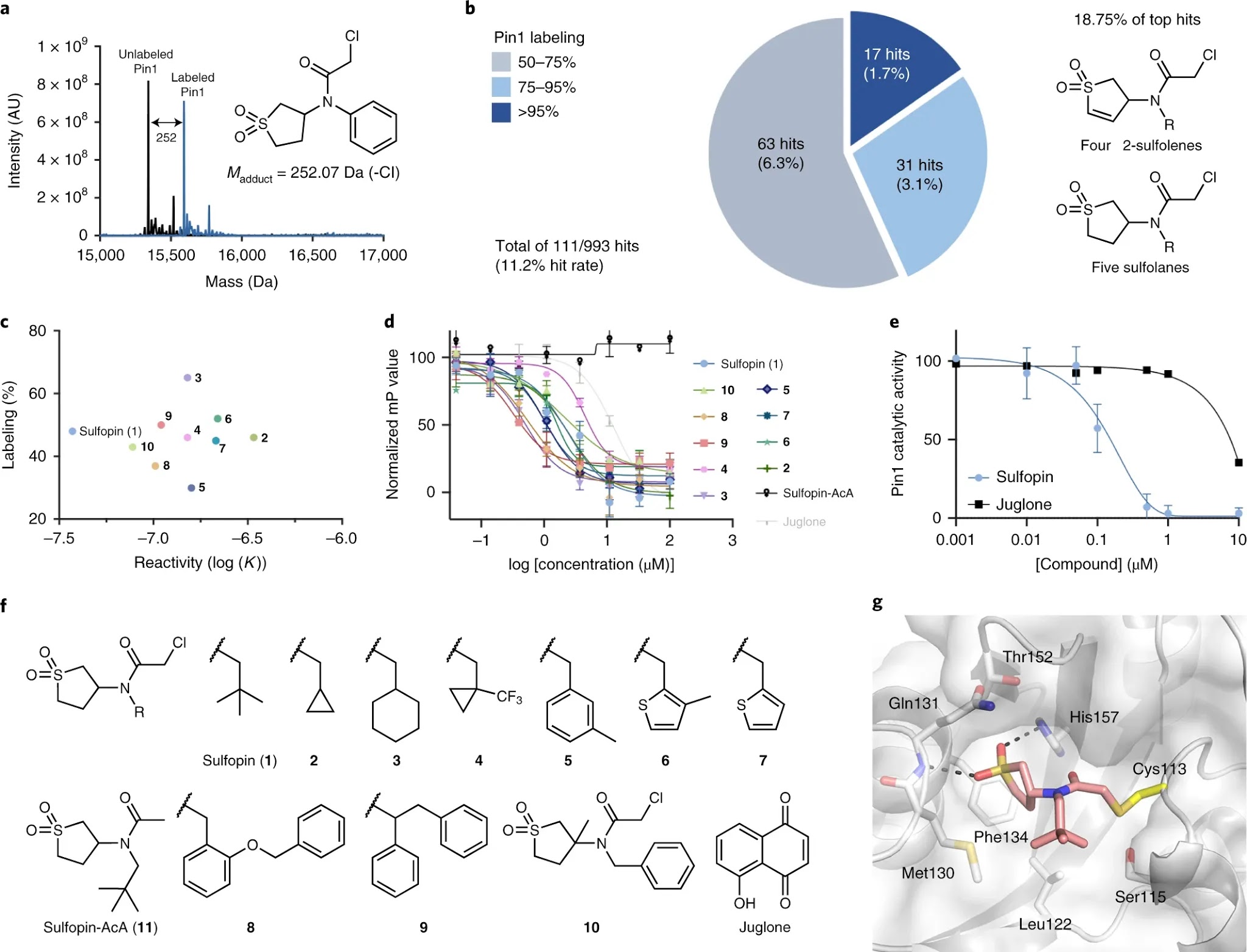
The peptidyl-prolyl isomerase, Pin1, is exploited in cancer to activate oncogenes and inactivate tumor suppressors. However, despite considerable efforts, Pin1 has remained an elusive drug target. Here, we screened an electrophilic fragment library to identify covalent inhibitors targeting Pin1’s active site Cys113, leading to the development of Sulfopin, a nanomolar Pin1 inhibitor. Sulfopin is highly selective, as validated by two independent chemoproteomics methods, achieves potent cellular and in vivo target engagement and phenocopies Pin1 genetic knockout. Pin1 inhibition had only a modest effect on cancer cell line viability. Nevertheless, Sulfopin induced downregulation of c-Myc target genes, reduced tumor progression and conferred survival benefit in murine and zebrafish models of MYCN-driven neuroblastoma, and in a murine model of pancreatic cancer. Our results demonstrate that Sulfopin is a chemical probe suitable for assessment of Pin1-dependent pharmacology in cells and in vivo, and that Pin1 warrants further investigation as a potential cancer drug target.