J. Am. Chem. Soc., 2018
DOI: 10.1021/jacs.8b09433
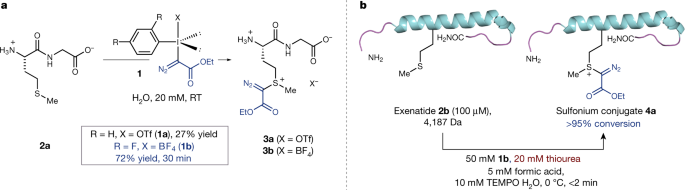
Olefin cross-metathesis (CM) is a viable reaction for the modification of alkene-containing proteins. Although allyl sulfide or selenide side-chain motifs in proteins can critically enhance the rate of CM reactions, no efficient method for their site-selective genetic incorporation into proteins has been reported to date. Here, through the systematic evaluation of olefin-bearing unnatural amino acids for their metabolic incorporation, we have discovered S-allylhomocysteine (Ahc) as a genetically encodable Met analogue that is not only processed by translational cellular machinery but also a privileged CM substrate residue in proteins. In this way, Ahc was used for efficient Met codon reassignment in a Met-auxotrophic strain of E. coli (B834 (DE3)) as well as metabolic labeling of protein in human cells and was reactive toward CM in several representative proteins. This expands the use of CM in the toolkit for “tag-and-modify” functionalization of proteins.