Kemper, E.K., Zhang, Y., Dix, M.M. & Benjamin F. Cravatt.
Nat Methods, 2022
https://www.nature.com/articles/s41592-022-01398-2
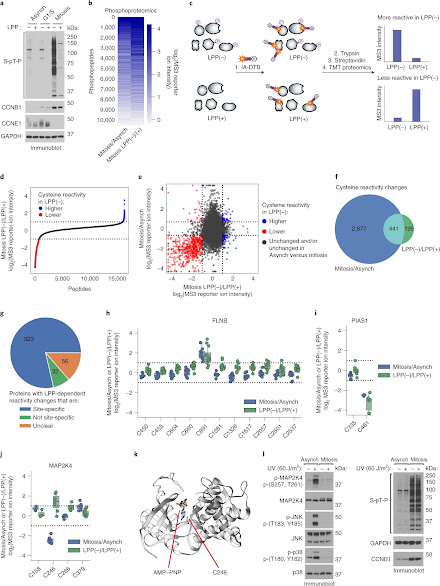
Proteomics has revealed that the ~20,000 human genes engender a far greater number of proteins, or proteoforms, that are diversified in large part by post-translational modifications (PTMs). How such PTMs affect protein structure and function is an active area of research but remains technically challenging to assess on a proteome-wide scale. Here, we describe a chemical proteomic method to quantitatively relate serine/threonine phosphorylation to changes in the reactivity of cysteine residues, a parameter that can affect the potential for cysteines to be post-translationally modified or engaged by covalent drugs. Leveraging the extensive high-stoichiometry phosphorylation occurring in mitotic cells, we discover numerous cysteines that exhibit phosphorylation-dependent changes in reactivity on diverse proteins enriched in cell cycle regulatory pathways. The discovery of bidirectional changes in cysteine reactivity often occurring in proximity to serine/threonine phosphorylation events points to the broad impact of phosphorylation on the chemical reactivity of proteins and the future potential to create small-molecule probes that differentially target proteoforms with PTMs.