Chem. Eur. J. 2022, e202201843
https://doi.org/10.1002/chem.202201843
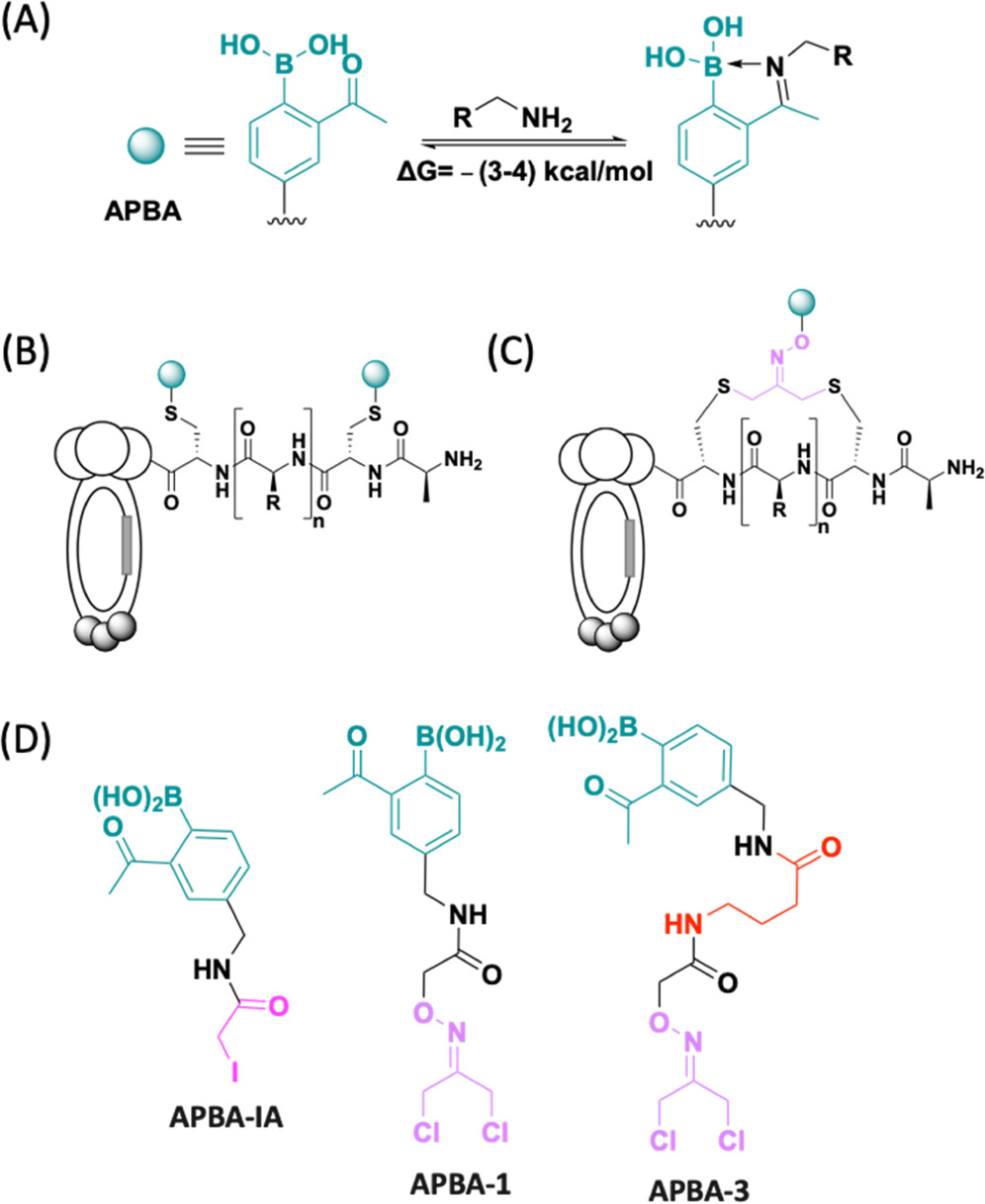
Cysteine bioconjugation serves as a powerful tool in biological research and has been widely used for the chemical modification of proteins, constructing antibody-drug conjugates, and enabling cell imaging studies. Cysteine conjugation reactions with fast kinetics and exquisite selectivity have been under heavy pursuit as they would allow clean protein modification with just stoichiometric amount of reagents, which minimizes side reaction, simplifies purification and broadens the functional group tolerance. In this concept, we summarize the recent advances of fast cysteine bioconjugation, and discuss the mechanism and chemical principles that underlie the high efficiencies of the newly developed cysteine reactive reagents.