Zhan Zhou, Xiaojuan Chen, Ying Fu, Ye Zhang, Shuyan Dai, Jun Li, Lin Chen, Guangyu Xu, Zhuchu Chen and Yongheng Chen
Chem. Commun., 2019
DOI: 10.1039/C9CC02052G
Biochemical and structural studies provide information on the mode of action of FGF401 as a selective, reversible covalent inhibitor of FGFR4. Kinase and proliferation assays reveal that FGF401 has the ability to overcome gatekeeper mutations in FGFR4.
Tuesday, April 30, 2019
Wednesday, April 24, 2019
Bioinspired Thiophosphorodichloridate Reagents for Chemoselective Histidine Bioconjugation
Shang Jia, Dan He, and Christopher J. Chang
Journal of the American Chemical Society, 2019
DOI: 10.1021/jacs.8b11912
Site-selective bioconjugation to native protein residues is a powerful tool for protein functionalization, with cysteine and lysine side chains being the most common points for attachment owing to their high nucleophilicity. We now report a strategy for histidine modification using thiophosphorodichloridate reagents that mimic post-translational histidine phosphorylation, enabling fast and selective labeling of protein histidines under mild conditions where various payloads can be introduced via copper-assisted alkyne–azide cycloaddition (CuAAC) chemistry. We establish that these reagents are particularly effective at covalent modification of His-tags, which are common motifs to facilitate protein purification, as illustrated by selective attachment of polyarginine cargoes to enhance the uptake of proteins into living cells. This work provides a starting point for probing and enhancing protein function using histidine-directed chemistry.
Tuesday, April 23, 2019
New Electrophiles and Strategies for Mechanism-Based and Targeted Covalent Inhibitor Design
Sneha Ray and Andrew S. Murkin
Biochemistry, 2019
DOI: 10.1021/acs.biochem.9b00293
Covalent inhibitors are experiencing a growing resurgence in drug design and are an increasingly useful tool in molecular biology. The ability to attach inhibitors to their targets by a covalent linkage offers pharmacodynamic and pharmacokinetic advantages, but this can also be a liability if undesired off-target reactions are not mitigated. The discovery of new electrophilic groups that react selectively with specific amino acid residues is therefore highly desirable in the design of targeted covalent inhibitors (TCIs). Additionally, the ability to control reactivity through exploitation of the target enzyme’s machinery, as in mechanism-based inhibitors (MBIs), greatly benefits from the discovery of new strategies. This Perspective showcases recent advances in electrophile development and their application in TCIs and MBIs exhibiting high selectivity for their targets.
Biochemistry, 2019
DOI: 10.1021/acs.biochem.9b00293
Covalent inhibitors are experiencing a growing resurgence in drug design and are an increasingly useful tool in molecular biology. The ability to attach inhibitors to their targets by a covalent linkage offers pharmacodynamic and pharmacokinetic advantages, but this can also be a liability if undesired off-target reactions are not mitigated. The discovery of new electrophilic groups that react selectively with specific amino acid residues is therefore highly desirable in the design of targeted covalent inhibitors (TCIs). Additionally, the ability to control reactivity through exploitation of the target enzyme’s machinery, as in mechanism-based inhibitors (MBIs), greatly benefits from the discovery of new strategies. This Perspective showcases recent advances in electrophile development and their application in TCIs and MBIs exhibiting high selectivity for their targets.
Friday, April 19, 2019
Covalent Inhibition in Drug Discovery
Ghosh, A. K., Samanta, I. , Mondal, A. and Liu, W. R.
ChemMedChem, 2019
doi:10.1002/cmdc.201900107
Although covalent inhibitors have been used as therapeutics for more than a century, there has been general resistance in the pharmaceutical industry against their further development due to safety concerns. This inclination has recently been reverted after the development of a wide variety of covalent inhibitors to address human health conditions along with the US Food and Drug Administration (FDA) approval of several covalent therapeutics for use in humans. Along with this exciting resurrection of an old drug discovery concept, this review surveys enzymes that can be targeted by covalent inhibitors for the treatment of human diseases. We focus on protein kinases, RAS proteins, and a few other enzymes that have been studied extensively as targets for covalent inhibition, with the aim to address challenges in designing effective covalent drugs and to provide suggestions in the area that have yet to be explored.
ChemMedChem, 2019
doi:10.1002/cmdc.201900107
Although covalent inhibitors have been used as therapeutics for more than a century, there has been general resistance in the pharmaceutical industry against their further development due to safety concerns. This inclination has recently been reverted after the development of a wide variety of covalent inhibitors to address human health conditions along with the US Food and Drug Administration (FDA) approval of several covalent therapeutics for use in humans. Along with this exciting resurrection of an old drug discovery concept, this review surveys enzymes that can be targeted by covalent inhibitors for the treatment of human diseases. We focus on protein kinases, RAS proteins, and a few other enzymes that have been studied extensively as targets for covalent inhibition, with the aim to address challenges in designing effective covalent drugs and to provide suggestions in the area that have yet to be explored.
Azabicyclic vinyl sulfones for residue-specific dual protein labelling
Chem. Sci., 2019, 10, 4515-4522
doi: 10.1039/C9SC00125E
We have developed [2.2.1]azabicyclic vinyl sulfone reagents that simultaneously enable cysteine-selective protein modification and introduce a handle for further bioorthogonal ligation. The reaction is fast and selective for cysteine relative to other amino acids that have nucleophilic side-chains, and the formed products are stable in human plasma and are moderately resistant to retro Diels–Alder degradation reactions. A model biotinylated [2.2.1]azabicyclic vinyl sulfone reagent was shown to efficiently label two cysteine-tagged proteins, ubiquitin and C2Am, under mild conditions (1–5 equiv. of reagent in NaPi pH 7.0, room temperature, 30 min). The resulting thioether-linked conjugates were stable and retained the native activity of the proteins. Finally, the dienophile present in the azabicyclic moiety on a functionalised C2Am protein could be fluorescently labelled through an inverse electron demand Diels–Alder reaction in cells to allow selective apoptosis imaging. The combined advantages of directness, site-specificity and easy preparation mean [2.2.1]azabicyclic vinyl sulfones can be used for residue-specific dual protein labelling/construction strategies with minimal perturbation of native function based simply on the attachment of an [2.2.1]azabicyclic moiety to cysteine.
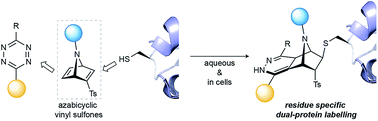
doi: 10.1039/C9SC00125E
We have developed [2.2.1]azabicyclic vinyl sulfone reagents that simultaneously enable cysteine-selective protein modification and introduce a handle for further bioorthogonal ligation. The reaction is fast and selective for cysteine relative to other amino acids that have nucleophilic side-chains, and the formed products are stable in human plasma and are moderately resistant to retro Diels–Alder degradation reactions. A model biotinylated [2.2.1]azabicyclic vinyl sulfone reagent was shown to efficiently label two cysteine-tagged proteins, ubiquitin and C2Am, under mild conditions (1–5 equiv. of reagent in NaPi pH 7.0, room temperature, 30 min). The resulting thioether-linked conjugates were stable and retained the native activity of the proteins. Finally, the dienophile present in the azabicyclic moiety on a functionalised C2Am protein could be fluorescently labelled through an inverse electron demand Diels–Alder reaction in cells to allow selective apoptosis imaging. The combined advantages of directness, site-specificity and easy preparation mean [2.2.1]azabicyclic vinyl sulfones can be used for residue-specific dual protein labelling/construction strategies with minimal perturbation of native function based simply on the attachment of an [2.2.1]azabicyclic moiety to cysteine.
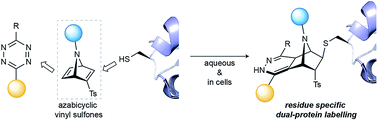
Monday, April 15, 2019
Leveraging Compound Promiscuity to Identify Targetable Cysteines within the Kinome
Suman Rao, Deepak Gurbani, Guangyan Du, Robert A Everley, Christopher M Browne, Apirat Chaikuad, Tan Li, Martin Schröder, Sudershan Gondi, Scott B Ficarro, Taebo Sim, Nam Doo Kim, Matthew J Berberich, Stefan Knapp, Jarrod A Marto, Kenneth D Westover, Peter K Sorger, Nathanael S Gray
Cell Chemical Biology, 2019
DOI: https://doi.org/10.1016/j.chembiol.2019.02.021
Covalent kinase inhibitors, which typically target cysteine residues, represent an important class of clinically relevant compounds. Approximately 215 kinases are known to have potentially targetable cysteines distributed across 18 spatially distinct locations proximal to the ATP-binding pocket. However, only 40 kinases have been covalently targeted, with certain cysteine sites being the primary focus. To address this disparity, we have developed a strategy that combines the use of a multi-targeted acrylamide-modified inhibitor, SM1-71, with a suite of complementary chemoproteomic and cellular approaches to identify additional targetable cysteines. Using this single multi-targeted compound, we successfully identified 23 kinases that are amenable to covalent inhibition including MKNK2, MAP2K1/2/3/4/6/7, GAK, AAK1, BMP2K, MAP3K7, MAPKAPK5, GSK3A/B, MAPK1/3, SRC, YES1, FGFR1, ZAK (MLTK), MAP3K1, LIMK1, and RSK2. The identification of nine of these kinases previously not targeted by a covalent inhibitor increases the number of targetable kinases and highlights opportunities for covalent kinase inhibitor development.
Cell Chemical Biology, 2019
DOI: https://doi.org/10.1016/j.chembiol.2019.02.021
Covalent kinase inhibitors, which typically target cysteine residues, represent an important class of clinically relevant compounds. Approximately 215 kinases are known to have potentially targetable cysteines distributed across 18 spatially distinct locations proximal to the ATP-binding pocket. However, only 40 kinases have been covalently targeted, with certain cysteine sites being the primary focus. To address this disparity, we have developed a strategy that combines the use of a multi-targeted acrylamide-modified inhibitor, SM1-71, with a suite of complementary chemoproteomic and cellular approaches to identify additional targetable cysteines. Using this single multi-targeted compound, we successfully identified 23 kinases that are amenable to covalent inhibition including MKNK2, MAP2K1/2/3/4/6/7, GAK, AAK1, BMP2K, MAP3K7, MAPKAPK5, GSK3A/B, MAPK1/3, SRC, YES1, FGFR1, ZAK (MLTK), MAP3K1, LIMK1, and RSK2. The identification of nine of these kinases previously not targeted by a covalent inhibitor increases the number of targetable kinases and highlights opportunities for covalent kinase inhibitor development.
Discovery of Covalent CDK14 Inhibitors with Pan-TAIRE Family Specificity
Fleur M Ferguson, Zainab M Doctor, Scott B Ficarro, Christopher M Browne, Jarrod A Marto, Jared L Johnson, Tomer M Yaron, Lewis C Cantley, Nam Doo Kim, Taebo Sim, Matthew J Berberich, Marian Kalocsay, Peter K Sorger, Nathanael S Gray
Cell Chemical Biology, 2019
DOI: https://doi.org/10.1016/j.chembiol.2019.02.015
Cyclin-dependent kinase 14 (CDK14) and other TAIRE family kinases (CDKs 15–18) are proteins that lack functional annotation but are frequent off-targets of clinical kinase inhibitors. In this study we develop and characterize FMF-04-159-2, a tool compound that specifically targets CDK14 covalently and possesses a TAIRE kinase-biased selectivity profile. This tool compound and its reversible analog were used to characterize the cellular consequences of covalent CDK14 inhibition, including an unbiased investigation using phospho-proteomics. To reduce confounding off-target activity, washout conditions were used to deconvolute CDK14-specific effects. This investigation suggested that CDK14 plays a supporting role in cell-cycle regulation, particularly mitotic progression, and identified putative CDK14 substrates. Together, these results represent an important step forward in understanding the cellular consequences of inhibiting CDK14 kinase activity.
Cell Chemical Biology, 2019
DOI: https://doi.org/10.1016/j.chembiol.2019.02.015
Cyclin-dependent kinase 14 (CDK14) and other TAIRE family kinases (CDKs 15–18) are proteins that lack functional annotation but are frequent off-targets of clinical kinase inhibitors. In this study we develop and characterize FMF-04-159-2, a tool compound that specifically targets CDK14 covalently and possesses a TAIRE kinase-biased selectivity profile. This tool compound and its reversible analog were used to characterize the cellular consequences of covalent CDK14 inhibition, including an unbiased investigation using phospho-proteomics. To reduce confounding off-target activity, washout conditions were used to deconvolute CDK14-specific effects. This investigation suggested that CDK14 plays a supporting role in cell-cycle regulation, particularly mitotic progression, and identified putative CDK14 substrates. Together, these results represent an important step forward in understanding the cellular consequences of inhibiting CDK14 kinase activity.
Thursday, April 11, 2019
Lysine-Targeted Inhibitors and Chemoproteomic Probes
Adolfo Cuesta and Jack Taunton
Annual Review of Biochemistry, 2019
DOI: https://www.annualreviews.org/doi/pdf/10.1146/annurev-biochem-061516-044805
Covalent inhibitors are widely used in drug discovery and chemical biology. Although covalent inhibitors are frequently designed to react with noncatalytic cysteines, many ligand binding sites lack an accessible cysteine. Here, we review recent advances in the chemical biology of lysine-targeted covalent inhibitors and chemoproteomic probes. By analyzing crystal structures of proteins bound to common metabolites and enzyme cofactors, we identify a large set of mostly unexplored lysines that are potentially targetable with covalent inhibitors. In addition, we describe mass spectrometry-based approaches for determining proteome-wide lysine ligandability and lysine-reactive chemoproteomic probes for assessing drug–target engagement. Finally, we discuss the design of amine-reactive inhibitors that form reversible covalent bonds with their protein targets.
keywords: aldehyde, sulfonyl fluoride, chemoproteomics, kinase, reversible covalent
Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors
Ruibin Liu, Zhi Yue, Cheng-Chieh Tsai, and Jana Shen
J. Am. Chem. Soc., 2019
DOI: 10.1021/jacs.8b13248
Targeted covalent inhibitor design is gaining increasing interest and acceptance. A typical covalent kinase inhibitor design targets a reactive cysteine; however, this strategy is limited due to the low abundance of cysteine and acquired drug resistance from point mutations. Inspired by the recent development of lysine-targeted chemical probes, we asked if nucleophilic (reactive) catalytic lysines are common based on the published crystal structures of the human kinome. Using a newly developed pKa prediction tool based on continuous constant pH molecular dynamics, the catalytic lysines of 8 unique kinases from various human kinase groups were retro- and prospectively predicted to be nucleophilic, when kinase is in the rare DFG-in/aC-out type of conformation. Importantly, other reactive lysines as well as cysteines at various locations were also identified. Based on the finding, we proposed a new strategy based on modification of selective type II reversible kinase inhibitors to discover highly selective, lysine-targeted covalent inhibitors. Traditional covalent drugs were discovered serendipitously; the presented tool, which can assess the reactivities of any potentially targetable residues, may assist and accelerate the rational discovery of new covalent inhibitors. Another significant finding of the work is that lysines and cysteines in kinases may adopt neutral and charged states at physiological pH, respectively. This finding may shift the current paradigm of computational studies of kinases, which assume standard protonation states.
J. Am. Chem. Soc., 2019
DOI: 10.1021/jacs.8b13248
Targeted covalent inhibitor design is gaining increasing interest and acceptance. A typical covalent kinase inhibitor design targets a reactive cysteine; however, this strategy is limited due to the low abundance of cysteine and acquired drug resistance from point mutations. Inspired by the recent development of lysine-targeted chemical probes, we asked if nucleophilic (reactive) catalytic lysines are common based on the published crystal structures of the human kinome. Using a newly developed pKa prediction tool based on continuous constant pH molecular dynamics, the catalytic lysines of 8 unique kinases from various human kinase groups were retro- and prospectively predicted to be nucleophilic, when kinase is in the rare DFG-in/aC-out type of conformation. Importantly, other reactive lysines as well as cysteines at various locations were also identified. Based on the finding, we proposed a new strategy based on modification of selective type II reversible kinase inhibitors to discover highly selective, lysine-targeted covalent inhibitors. Traditional covalent drugs were discovered serendipitously; the presented tool, which can assess the reactivities of any potentially targetable residues, may assist and accelerate the rational discovery of new covalent inhibitors. Another significant finding of the work is that lysines and cysteines in kinases may adopt neutral and charged states at physiological pH, respectively. This finding may shift the current paradigm of computational studies of kinases, which assume standard protonation states.
Saturday, April 6, 2019
Characterising Covalent Warhead Reactivity
James S. Martin, Claire J. MacKenzie, Daniel Fletcher, Ian H. Gilbert,
Bioorganic & Medicinal Chemistry, 2019, DOI: 10.1016/j.bmc.2019.04.002
Many drugs currently used are covalent inhibitors and irreversibly inhibit their targets. Most of these were discovered through serendipity. Covalent inhibitions can have many advantages from a pharmacokinetic perspective. However, until recently most organisations have shied away from covalent compound design due to fears of non-specific inhibition of off-target proteins leading to toxicity risks. However, there has been a renewed interest in covalent modifiers as potential drugs, as it possible to get highly selective compounds. It is therefore important to know how reactive a warhead is and to be able to select the least reactive warhead possible to avoid toxicity. A robust NMR based assay was developed and used to measure the reactivity of a variety of covalent warheads against serine and cysteine - the two most common targets for covalent drugs. A selection of these warheads also had their reactivity measured against threonine, tyrosine, lysine, histidine and arginine to better understand our ability to target non-traditional residues. The reactivity was also measured at various pHs to assess what effect the environment in the active site would have on these reactions. The reactivity of a covalent modifier was found to be very dependent on the residue.

Bioorganic & Medicinal Chemistry, 2019, DOI: 10.1016/j.bmc.2019.04.002
Many drugs currently used are covalent inhibitors and irreversibly inhibit their targets. Most of these were discovered through serendipity. Covalent inhibitions can have many advantages from a pharmacokinetic perspective. However, until recently most organisations have shied away from covalent compound design due to fears of non-specific inhibition of off-target proteins leading to toxicity risks. However, there has been a renewed interest in covalent modifiers as potential drugs, as it possible to get highly selective compounds. It is therefore important to know how reactive a warhead is and to be able to select the least reactive warhead possible to avoid toxicity. A robust NMR based assay was developed and used to measure the reactivity of a variety of covalent warheads against serine and cysteine - the two most common targets for covalent drugs. A selection of these warheads also had their reactivity measured against threonine, tyrosine, lysine, histidine and arginine to better understand our ability to target non-traditional residues. The reactivity was also measured at various pHs to assess what effect the environment in the active site would have on these reactions. The reactivity of a covalent modifier was found to be very dependent on the residue.

Subscribe to:
Posts (Atom)