Grob, N. M.; Remarcik, C.; Rössler, S. L.; Wong, J. Y. K.; Wang, J. C. K.; Tao, J.; Smith, C. L.; Loas, A.; Buchwald, S. L.; Eaton, D. L.; Preciado López, M.; Pentelute, B. L.
ChemRxiv 2023.
https://doi.org/10.26434/chemrxiv-2023-hvq1k
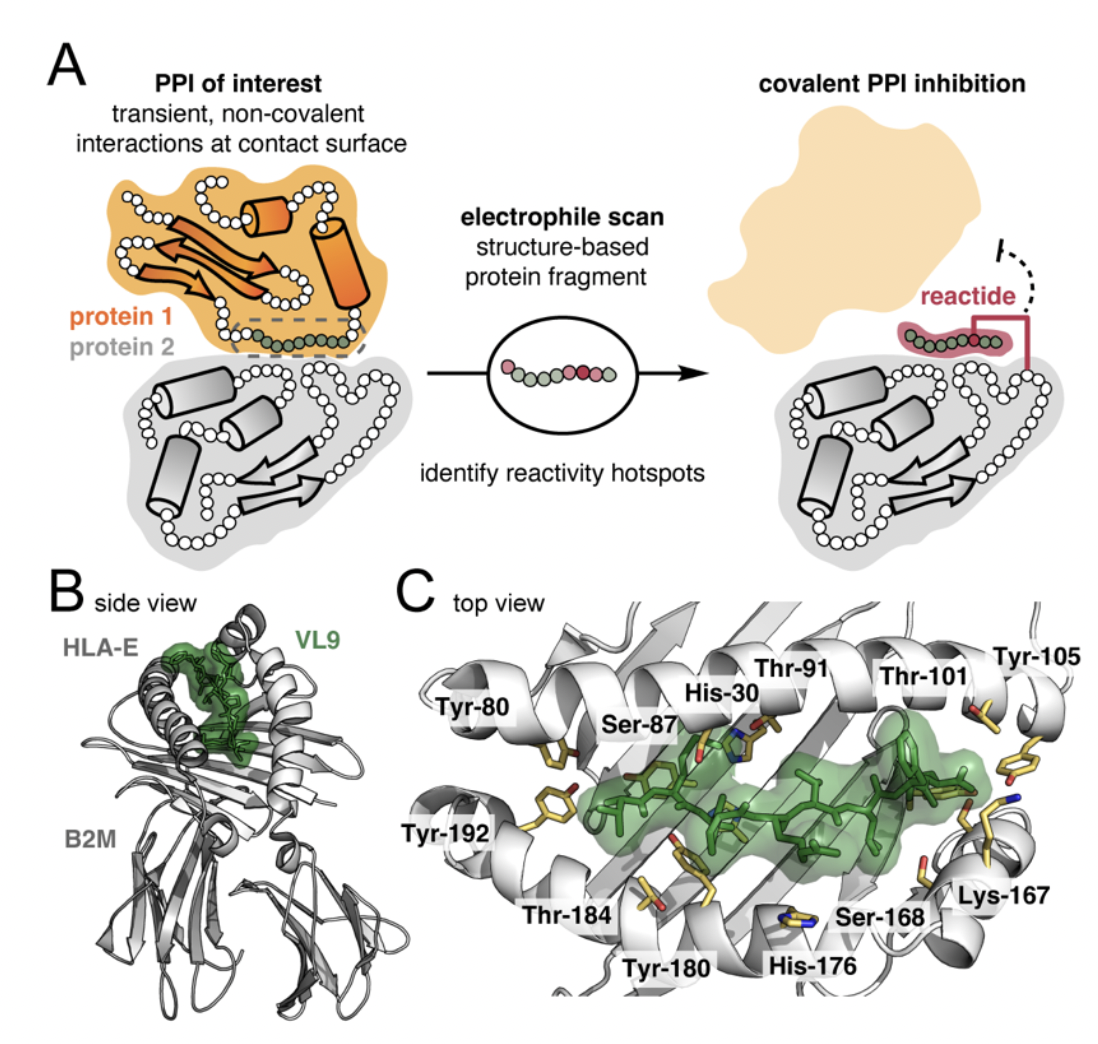
Protein–protein interactions (PPIs) are intriguing targets in drug discovery and development. Peptides are well suited to target PPIs, which typically present with large surface areas lacking distinct features and deep binding pockets. To improve binding interactions to these topologies by PPI-focused therapeutics and advance their development, potential ligands can be equipped with electrophilic groups to enable binding through covalent mechanisms of action. We report a strategy termed electrophile scanning to identify reactivity hotspots in a known peptide ligand. Cysteine mutants of the ligand are used to install protein-reactive modifiers via a palladium oxidative addition complex (Pd-OAC). Reactivity hotspots are revealed by cross-linking reactions with the target protein under physiological conditions. In a system with the 9-mer peptide antigen VL9 and MHC class I receptor HLA-E, we identify two reactivity hotspots that afford up to 87% conversion to the protein–peptide conjugate within 4 hours. The reactions are specific to the target protein in vitro and dependent on the peptide sequence. Moreover, the cross-linked peptide successfully inhibits molecular recognition of HLA-E by CD94─NKG2A possibly due to structural changes enacted at the PPI interface. The results illustrate the potential of electrophile scanning as a tool for rapid discovery and development of covalent peptide binders.