Xiaoyun Lu, Jeff Bruce Smaill, and Ke Ding
J. Med. Chem. 2020
https://doi.org/10.1021/acs.jmedchem.0c00507
Clinically acquired resistance to small molecule kinase inhibitors (SMKIs) has become a major “unmet clinical need” in cancer therapy. To date, there are six SMKIs to be approved for the treatment of cancer patients through targeting of clinically acquired resistance caused by on-target mutations, these are mainly focused on the mutant kinases Bcr-Abl T315I, EGFR T790M and ALK L1196M. Herein, we summarize the major medicinal chemistry strategies employed in the discovery of these representative SMKIs, such as avoiding steric hindrance, making additional interactions with mutated residues and forming a covalent bond with an active site cysteine to override resistance observed for reversible inhibitors. Additionally, we also briefly describe allosteric kinase inhibitors and proteolysis targeting chimera (PROTAC) as two other potential strategies, while addressing future opportunities in this area.
Sunday, May 31, 2020
Thursday, May 28, 2020
Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease
Alice Douangamath, Daren Fearon, Paul Gehrtz, Tobias Krojer, Petra Lukacik, C. David Owen, Efrat Resnick, Claire Strain-Damerell, Anthony aimon, Péter Ábrányi-Balogh, Jose Brandaõ-Neto, Anna Carberry, Gemma Davison, Alexandre Dias, Thomas D Downes, Louise Dunnett, Michael Fairhead, James D Firth, S. Paul Jones, Aaron Keely, György M Keserü, Hanna F Klein, Matthew P Martin, Martin E.M. Noble, Peter O'Brien, Ailsa Powell, Rambabu Reddi, Rachael Skyner, Matthew Snee, Michael J Waring, Conor Wild, Nir London, Frank von Delft, Martin A Walsh
bioRxiv 2020; doi: https://doi.org/10.1101/2020.05.27.118117
COVID-19, caused by SARS-CoV-2, lacks effective therapeutics. Additionally, no antiviral drugs or vaccines were developed against the closely related coronavirus, SARS-CoV-1 or MERS-CoV, despite previous zoonotic outbreaks. To identify starting points for such therapeutics, we performed a large-scale screen of electrophile and non-covalent fragments through a combined mass spectrometry and X-ray approach against the SARS-CoV-2 main protease, one of two cysteine viral proteases essential for viral replication. Our crystallographic screen identified 71 hits that span the entire active site, as well as 3 hits at the dimer interface. These structures reveal routes to rapidly develop more potent inhibitors through merging of covalent and non-covalent fragment hits; one series of low-reactivity, tractable covalent fragments was progressed to discover improved binders. These combined hits offer unprecedented structural and reactivity information for on-going structure-based drug design against SARS-CoV-2 main protease.

bioRxiv 2020; doi: https://doi.org/10.1101/2020.05.27.118117
COVID-19, caused by SARS-CoV-2, lacks effective therapeutics. Additionally, no antiviral drugs or vaccines were developed against the closely related coronavirus, SARS-CoV-1 or MERS-CoV, despite previous zoonotic outbreaks. To identify starting points for such therapeutics, we performed a large-scale screen of electrophile and non-covalent fragments through a combined mass spectrometry and X-ray approach against the SARS-CoV-2 main protease, one of two cysteine viral proteases essential for viral replication. Our crystallographic screen identified 71 hits that span the entire active site, as well as 3 hits at the dimer interface. These structures reveal routes to rapidly develop more potent inhibitors through merging of covalent and non-covalent fragment hits; one series of low-reactivity, tractable covalent fragments was progressed to discover improved binders. These combined hits offer unprecedented structural and reactivity information for on-going structure-based drug design against SARS-CoV-2 main protease.

Thursday, May 21, 2020
ABHD17 enzymes regulate dynamic plasma membrane palmitoylation and N-Ras-dependent cancer growth [@MJNiphakis]
Jarrett R Remsberg, Radu M Suciu, Noemi A Zambetti, Thomas W Hanigan, Ari J Firestone, Anagha Inguva, Amy Long, Nhi Ngo, Kenneth M Lum, Cassandra L Henry, Stewart K Richardson, Marina Predovic, Ben Huang, Amy R Howell, Micah J Niphakis, Kevin Shannon, Benjamin F Cravatt
bioRxiv, 2020
doi: https://doi.org/10.1101/2020.05.21.108316
A subset of Ras proteins, including N-Ras, depend on a palmitoylation/depalmitoylation cycle to regulate their subcellular trafficking and oncogenicity. General lipase inhibitors such as Palmostatin M block N-Ras depalmitoylation, but lack specificity and target several enzymes displaying depalmitoylase activity. Here, we describe ABD957, a potent and selective covalent inhibitor of the ABHD17 family of depalmitoylases, and show that this compound impairs N-Ras depalmitoylation in human acute myeloid leukemia (AML) cells. ABD957 produced partial effects on N-Ras palmitoylation compared to Palmostatin M, but was much more selective across the proteome, reflecting a plasma membrane-delineated action on dynamically palmitoylated proteins. Finally, ABD957 impaired N-Ras signaling and the growth of NRAS-mutant AML cells in a manner that synergizes with MEK inhibition. Our findings uncover a surprisingly restricted role for ABHD17 enzymes in modulating the N-Ras palmitoylation cycle and suggest that ABHD17 inhibitors may have value as targeted therapies for NRAS-mutant cancers

 .
.
bioRxiv, 2020
doi: https://doi.org/10.1101/2020.05.21.108316
A subset of Ras proteins, including N-Ras, depend on a palmitoylation/depalmitoylation cycle to regulate their subcellular trafficking and oncogenicity. General lipase inhibitors such as Palmostatin M block N-Ras depalmitoylation, but lack specificity and target several enzymes displaying depalmitoylase activity. Here, we describe ABD957, a potent and selective covalent inhibitor of the ABHD17 family of depalmitoylases, and show that this compound impairs N-Ras depalmitoylation in human acute myeloid leukemia (AML) cells. ABD957 produced partial effects on N-Ras palmitoylation compared to Palmostatin M, but was much more selective across the proteome, reflecting a plasma membrane-delineated action on dynamically palmitoylated proteins. Finally, ABD957 impaired N-Ras signaling and the growth of NRAS-mutant AML cells in a manner that synergizes with MEK inhibition. Our findings uncover a surprisingly restricted role for ABHD17 enzymes in modulating the N-Ras palmitoylation cycle and suggest that ABHD17 inhibitors may have value as targeted therapies for NRAS-mutant cancers


Wednesday, May 20, 2020
Structure and Characterization of a Covalent Inhibitor of Src Kinase [@kenwestover]
Gurbani Deepak, Du Guangyan, Henning Nathaniel J., Rao Suman, Bera Asim K., Zhang Tinghu, Gray Nathanael S., Westover Kenneth D.
Front. Mol. Biosci., 2020
DOI: https://doi.org/10.3389/fmolb.2020.00081
Unregulated Src activity promotes malignant processes in cancer, but no Src-directed targeted therapies are used clinically, possibly because early Src inhibitors produce off-target effects leading to toxicity. Improved selective Src inhibitors may enable Src-directed therapies. Previously, we reported an irreversible Src inhibitor, DGY-06-116, based on the hybridization of dasatinib and a promiscuous covalent kinase probe SM1-71. Here, we report biochemical and biophysical characterization of this compound. An x-ray co-crystal structure of DGY-06-116: Src shows a covalent interaction with the kinase p-loop and occupancy of the back hydrophobic kinase pocket, explaining its high potency, and selectivity. However, a reversible analog also shows similar potency. Kinetic analysis shows a slow inactivation rate compared to other clinically approved covalent kinase inhibitors, consistent with a need for p-loop movement prior to covalent bond formation. Overall, these results suggest that a strong reversible interaction is required to allow sufficient time for the covalent reaction to occur. Further optimization of the covalent linker may improve the kinetics of covalent bond formation.

Front. Mol. Biosci., 2020
DOI: https://doi.org/10.3389/fmolb.2020.00081
Unregulated Src activity promotes malignant processes in cancer, but no Src-directed targeted therapies are used clinically, possibly because early Src inhibitors produce off-target effects leading to toxicity. Improved selective Src inhibitors may enable Src-directed therapies. Previously, we reported an irreversible Src inhibitor, DGY-06-116, based on the hybridization of dasatinib and a promiscuous covalent kinase probe SM1-71. Here, we report biochemical and biophysical characterization of this compound. An x-ray co-crystal structure of DGY-06-116: Src shows a covalent interaction with the kinase p-loop and occupancy of the back hydrophobic kinase pocket, explaining its high potency, and selectivity. However, a reversible analog also shows similar potency. Kinetic analysis shows a slow inactivation rate compared to other clinically approved covalent kinase inhibitors, consistent with a need for p-loop movement prior to covalent bond formation. Overall, these results suggest that a strong reversible interaction is required to allow sufficient time for the covalent reaction to occur. Further optimization of the covalent linker may improve the kinetics of covalent bond formation.

Tuesday, May 19, 2020
Identification of a Covalent Molecular Inhibitor of Anti-apoptotic BFL-1 by Disulfide Tethering
Edward P.Harvey, Zachary J. Hauseman, Daniel T. Cohen, T. Justin Rettenmaier, Susan Lee, Annissa J.Huhn, Thomas E. Wales, Hyuk-Soo Seo, James Luccarelli1, Catherine E.Newman, Rachel M.Guerra, Gregory H.Bird, Sirano Dhe-Paganon, John R.Engen, James A.Wells, Loren D. Walensky
Cell Chemical Biology, 2020
The BCL-2 family is composed of anti- and pro-apoptotic members that respectively protect or disrupt mitochondrial integrity. Anti-apoptotic overexpression can promote oncogenesis by trapping the BCL-2 homology 3 (BH3) “killer domains” of pro-apoptotic proteins in a surface groove, blocking apoptosis. Groove inhibitors, such as the relatively large BCL-2 drug venetoclax (868 Da), have emerged as cancer therapies. BFL-1 remains an undrugged oncogenic protein and can cause venetoclax resistance. Having identified a unique C55 residue in the BFL-1 groove, we performed a disulfide tethering screen to determine if C55 reactivity could enable smaller molecules to block BFL-1's BH3-binding functionality. We found that a disulfide-bearing N-acetyltryptophan analog (304 Da adduct) effectively targeted BFL-1 C55 and reversed BFL-1-mediated suppression of mitochondrial apoptosis. Structural analyses implicated the conserved leucine-binding pocket of BFL-1 as the interaction site, resulting in conformational remodeling. Thus, therapeutic targeting of BFL-1 may be achievable through the design of small, cysteine-reactive drugs.

Alkynyl Benzoxazines and Dihydroquinazolines as Cysteine Targeting Covalent Warheads and Their Application in Identification of Selective Irreversible Kinase Inhibitors
Kirsten McAulay, Emily A Hoyt, Morgan Thomas, Marianne Schimpl, Michael Steven Bodnarchuk, Hilary Jane Lewis, Derek Barratt, Deepa Bhavsar, David M. Robinson, Michael J. Deery, Derek Ogg, Gonçalo J.L. Bernardes, Richard A Ward, Michael J. Waring, and Jason Grant Kettle
Journal of the American Chemical Society 2020
DOI: 10.1021/jacs.9b13391
With a resurgence in interest in covalent drugs, there is need to identify new moieties capable of cysteine bond formation that are differentiated from commonly employed systems such as acrylamide. Herein, we report on the discovery of new alkynyl benzoxazine and dihydroquinazoline moieties capable of covalent reaction with cysteine. Their utility as alternative electrophilic warheads for chemical biological probes and drug molecules is demonstrated through site-selective protein modification and incorporation into kinase drug scaffolds. A potent covalent inhibitor of JAK3 kinase was identified with superior selectivity across the kinome and improvements in in vitro pharmacokinetic profile relative to the related acrylamide-based inhibitor. In addition, the use of a novel heterocycle as cysteine reactive warhead is employed to target Cys788 in c-KIT where acrylamide has previously failed to form covalent interactions. These new reactive and selective heterocyclic warheads supplement the current repertoire for cysteine covalent modification whilst avoiding some of the limitations generally associated with established moieties.
Journal of the American Chemical Society 2020
DOI: 10.1021/jacs.9b13391
With a resurgence in interest in covalent drugs, there is need to identify new moieties capable of cysteine bond formation that are differentiated from commonly employed systems such as acrylamide. Herein, we report on the discovery of new alkynyl benzoxazine and dihydroquinazoline moieties capable of covalent reaction with cysteine. Their utility as alternative electrophilic warheads for chemical biological probes and drug molecules is demonstrated through site-selective protein modification and incorporation into kinase drug scaffolds. A potent covalent inhibitor of JAK3 kinase was identified with superior selectivity across the kinome and improvements in in vitro pharmacokinetic profile relative to the related acrylamide-based inhibitor. In addition, the use of a novel heterocycle as cysteine reactive warhead is employed to target Cys788 in c-KIT where acrylamide has previously failed to form covalent interactions. These new reactive and selective heterocyclic warheads supplement the current repertoire for cysteine covalent modification whilst avoiding some of the limitations generally associated with established moieties.
Monday, May 18, 2020
In vivo imaging of the tumor‐associated enzyme NCEH1 with a covalent PET probe
Chang, J., Bhuiyan, M., Tsai, H., Zhang, H., Li, G., Fathi, S., McCutcheon, D., Leoni, L., Freifelder, R., Chen, C. and Moellering, R.
Angew. Chem. Int. Ed. 2020
doi:10.1002/anie.202004762
Here we report the development of an 18F‐labeled, activity‐based small molecule probe targeting the cancer‐associated serine hydrolase NCEH1. We undertook a focused medicinal chemistry campaign to simultaneously preserve potent and specific NCEH1 labeling in live cells and animals, while permitting facile 18F radionuclide incorporation required for PET imaging. The resulting molecule, [18F]JW199, labels active NCEH1 in live cells at nM concentrations and greater than 1,000‐fold selectivity relative to other serine hydrolases. [18F]JW199 displays rapid, NCEH1‐dependent accumulation in mouse tissues. Finally, we demonstrate that [18F]JW199 labels aggressive cancer tumor cells in vivo, which uncovered localized NCEH1 activity at the leading edge of triple‐negative breast cancer tumors, suggesting roles for NCEH1 in tumor aggressiveness and metastasis. More generally, these data support the broader development of potent and specific covalent PET probes to visualize localized, active enzymes in live animals.

Angew. Chem. Int. Ed. 2020
doi:10.1002/anie.202004762
Here we report the development of an 18F‐labeled, activity‐based small molecule probe targeting the cancer‐associated serine hydrolase NCEH1. We undertook a focused medicinal chemistry campaign to simultaneously preserve potent and specific NCEH1 labeling in live cells and animals, while permitting facile 18F radionuclide incorporation required for PET imaging. The resulting molecule, [18F]JW199, labels active NCEH1 in live cells at nM concentrations and greater than 1,000‐fold selectivity relative to other serine hydrolases. [18F]JW199 displays rapid, NCEH1‐dependent accumulation in mouse tissues. Finally, we demonstrate that [18F]JW199 labels aggressive cancer tumor cells in vivo, which uncovered localized NCEH1 activity at the leading edge of triple‐negative breast cancer tumors, suggesting roles for NCEH1 in tumor aggressiveness and metastasis. More generally, these data support the broader development of potent and specific covalent PET probes to visualize localized, active enzymes in live animals.

Thursday, May 14, 2020
Covalent Immune Recruiters: Tools to Gain Chemical Control Over Immune Recognition
Lake B, Serniuck N, Kapcan E, Wang A, Rullo AF.
ACS Chemical Biology. 2020.
https://doi.org/10.1021/acschembio.0c00112
Unprecedented progress made in the treatment of cancer using the body’s own immune system has encouraged the development of synthetic molecule based immunotherapeutics. An emerging class of these compounds, called Antibody Recruiting Molecules (ARMs) or Antibody Engagers (AEs), functions by reversibly binding antibodies naturally present in human serum and recruiting these to cancer cells. The recruited antibodies then engage immune cells to form quaternary complexes that drive cancer eradication. Despite their promise, the requirement to form quaternary complexes governed by multiple equilibria complicates an understanding of their in vivo efficacy. Particularly problematic are low endogenous serum antibody concentrations and rapid clearance of AEs from circulation. Here we describe a new class of trifunctional chemical tools we call covalent immune recruiters (CIRs). CIRs covalently label specific serum antibodies in a selective manner with a target protein binding ligand. CIRs thereby exert well-defined control over antibody recruitment and simplify quaternary complex equilibrium, enabling probing of the resultant effects on immune recognition. We demonstrate CIRs can selectively covalently label anti-DNP IgG, a natural human antibody, directly in human serum to drive efficient immune cell recognition of targets. We expect CIRs will be useful tools to probe how quaternary complex stability impacts the immune recognition of cancer in vivo, revealing new design principles to guide the development of future AEs.

ACS Chemical Biology. 2020.
https://doi.org/10.1021/acschembio.0c00112
Unprecedented progress made in the treatment of cancer using the body’s own immune system has encouraged the development of synthetic molecule based immunotherapeutics. An emerging class of these compounds, called Antibody Recruiting Molecules (ARMs) or Antibody Engagers (AEs), functions by reversibly binding antibodies naturally present in human serum and recruiting these to cancer cells. The recruited antibodies then engage immune cells to form quaternary complexes that drive cancer eradication. Despite their promise, the requirement to form quaternary complexes governed by multiple equilibria complicates an understanding of their in vivo efficacy. Particularly problematic are low endogenous serum antibody concentrations and rapid clearance of AEs from circulation. Here we describe a new class of trifunctional chemical tools we call covalent immune recruiters (CIRs). CIRs covalently label specific serum antibodies in a selective manner with a target protein binding ligand. CIRs thereby exert well-defined control over antibody recruitment and simplify quaternary complex equilibrium, enabling probing of the resultant effects on immune recognition. We demonstrate CIRs can selectively covalently label anti-DNP IgG, a natural human antibody, directly in human serum to drive efficient immune cell recognition of targets. We expect CIRs will be useful tools to probe how quaternary complex stability impacts the immune recognition of cancer in vivo, revealing new design principles to guide the development of future AEs.
A Fast and Clean BTK Inhibitor [@london_lab]
Ronen Gabizon, Nir London
J. Med. Chem. 2020
https://doi.org/10.1021/acs.jmedchem.0c00597
Bruton’s tyrosine kinase (BTK) is a major drug target for B-cell related malignancies; however, existing BTK inhibitors approved for cancer treatment have significant off-targets that limit their use for autoimmune and inflammatory diseases. Remibrutinib (LOU064) is a novel covalent BTK inhibitor that binds an inactive BTK conformation, which affords it unprecedented selectivity. Its optimization led to rapid BTK engagement in vivo and fast clearance, further limiting systemic exposure. Remibrutinib is currently in phase 2 clinical trials for treatment of chronic urticaria and Sjoegren’s syndrome.

J. Med. Chem. 2020
https://doi.org/10.1021/acs.jmedchem.0c00597
Bruton’s tyrosine kinase (BTK) is a major drug target for B-cell related malignancies; however, existing BTK inhibitors approved for cancer treatment have significant off-targets that limit their use for autoimmune and inflammatory diseases. Remibrutinib (LOU064) is a novel covalent BTK inhibitor that binds an inactive BTK conformation, which affords it unprecedented selectivity. Its optimization led to rapid BTK engagement in vivo and fast clearance, further limiting systemic exposure. Remibrutinib is currently in phase 2 clinical trials for treatment of chronic urticaria and Sjoegren’s syndrome.

Wednesday, May 13, 2020
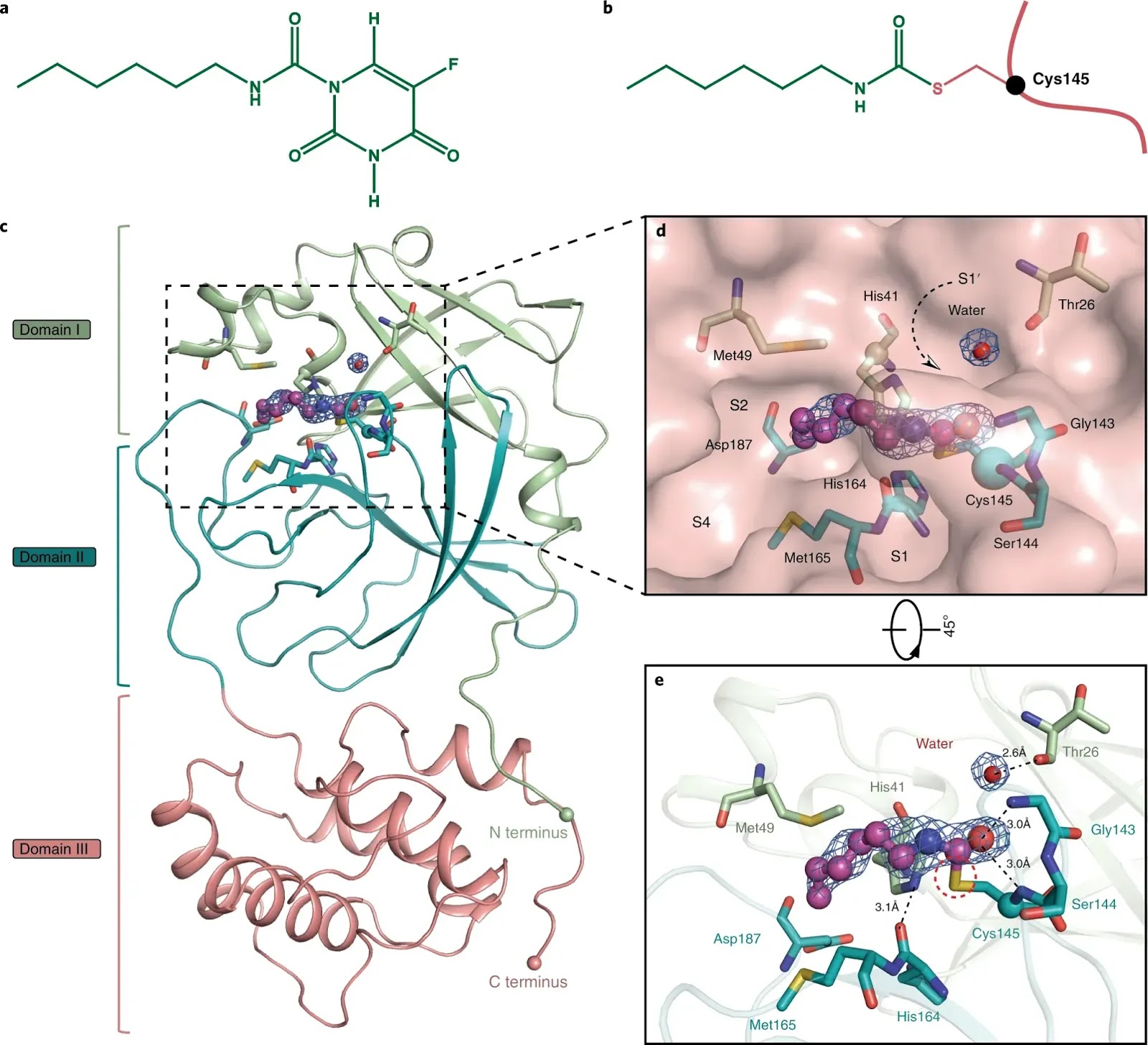
Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur
Zhenming Jin, Yao Zhao, Yuan Sun, Bing Zhang, Haofeng Wang, Yan Wu, Yan Zhu, Chen Zhu, Tianyu Hu, Xiaoyu Du, Yinkai Duan, Jing Yu, Xiaobao Yang, Xiuna Yang, Kailin Yang, Xiang Liu, Luke W. Guddat, Gengfu Xiao, Leike Zhang, Haitao Yang & Zihe Rao
Nat Struct Mol Biol, 2020
https://doi.org/10.1038/s41594-020-0440-6

Nat Struct Mol Biol, 2020
https://doi.org/10.1038/s41594-020-0440-6
The antineoplastic drug carmofur is shown to inhibit the SARS-CoV-2 main protease (Mpro). Here, the X-ray crystal structure of Mpro in complex with carmofur reveals that the carbonyl reactive group of carmofur is covalently bound to catalytic Cys145, whereas its fatty acid tail occupies the hydrophobic S2 subsite. Carmofur inhibits viral replication in cells (EC50 = 24.30 μM) and is a promising lead compound to develop new antiviral treatment for COVID-19.
Covalent Targeting of Ras G12C by Rationally Designed Peptidomimetics
Daniel Y Yoo, Andrew D. Hauser, Stephen T Joy, Dafna Bar-Sagi, and Paramjit S. Arora
ACS Chemical Biology 2020
DOI: 10.1021/acschembio.0c00204
Protein-protein interactions (PPIs) play a critical role in fundamental biological processes. Competitive inhibition of these interfaces requires compounds that can access discontinuous binding epitopes along a large, shallow binding surface area. Conformationally- defined protein surface mimics present a viable route to target these interactions. However, the development of minimal protein mimics that engage intracellular targets with high affinity remains a major challenge because mimicry of a portion of the binding interface is often associated with the loss of critical binding interactions. Covalent targeting provides an attractive approach to overcome the loss of non-covalent contacts but have the inherent risk of dominating non-covalent contacts and increasing the likelihood of non-selective binding. Here, we report the iterative design of a proteolytically-stable helix mimic that covalently targets oncogenic G12C Ras as a model system. We explored several electrophiles to optimize preferential alkylation with the desired C12 on Ras. The designed lead peptide modulates nucleotide exchange, inhibits activation of the Ras-mediated signalling cascade, and is selectively toxic towards mutant G12C Ras cancer cells. The relatively high frequency of acquired cysteines as missense mutations in cancer and other diseases suggests that covalent peptides may offer an untapped therapeutic approach for targeting aberrant protein interactions.

ACS Chemical Biology 2020
DOI: 10.1021/acschembio.0c00204
Protein-protein interactions (PPIs) play a critical role in fundamental biological processes. Competitive inhibition of these interfaces requires compounds that can access discontinuous binding epitopes along a large, shallow binding surface area. Conformationally- defined protein surface mimics present a viable route to target these interactions. However, the development of minimal protein mimics that engage intracellular targets with high affinity remains a major challenge because mimicry of a portion of the binding interface is often associated with the loss of critical binding interactions. Covalent targeting provides an attractive approach to overcome the loss of non-covalent contacts but have the inherent risk of dominating non-covalent contacts and increasing the likelihood of non-selective binding. Here, we report the iterative design of a proteolytically-stable helix mimic that covalently targets oncogenic G12C Ras as a model system. We explored several electrophiles to optimize preferential alkylation with the desired C12 on Ras. The designed lead peptide modulates nucleotide exchange, inhibits activation of the Ras-mediated signalling cascade, and is selectively toxic towards mutant G12C Ras cancer cells. The relatively high frequency of acquired cysteines as missense mutations in cancer and other diseases suggests that covalent peptides may offer an untapped therapeutic approach for targeting aberrant protein interactions.

Tuesday, May 12, 2020
An Azidoribose Probe to Track Ketoamine Adducts in Histone Ribose Glycation [@David_Lab_MSK]
Igor Maksimovic, Qingfei Zheng, Marissa N. Trujillo, James J. Galligan, and Yael David
Journal of the American Chemical Society 2020
DOI: 10.1021/jacs.0c01325
Journal of the American Chemical Society 2020
DOI: 10.1021/jacs.0c01325
Reactive cellular metabolites can modify macromolecules and form adducts known as non-enzymatic covalent modifications (NECMs). Dissecting the mechanisms, regulation and consequences of NECMs, such as glycation, has been challenging due to the complex and often ambiguous nature of the adducts formed. Directly tracking the formation of modifications on key targets to uncover their underlying physiological importance requires specific chemical tools. Here we present the novel chemoenzymatic syntheses of an active azido-modified ribose analog, 5-azidoribose (5-AR), as well as an inactive control derivative, 1-azidoribose (1-AR) and their application towards understanding protein ribose-glycation in vitro and in cellulo. With these new probes we found that, similar to MGO-glycation, ribose glycation specifically accumulates on histones. In addition to fluorescent labeling, we demonstrate the utility of the probe in enriching modified targets, which were identified by label-free quantitative proteomics and high-resolution MS/MS workflows. Finally, we establish that the known oncoprotein and hexose deglycase, fructosamine 3-kinase (FN3K), recognizes and facilitates the removal of 5-AR glycation adducts in live cells, supporting the dynamic regulation of ribose glycation as well as validating the probe as a new chemical tool to monitor FN3K activity. Altogether, we demonstrate this probe’s utilities to uncover ribose-glycation and deglycation events as well as tracking FN3K activity towards establishing its potential as a new cancer vulnerability.
Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles
Ned A. Porter, Libin Xu, and Derek A. Pratt
Chemistry 2020, 2(2), 390-417
https://doi.org/10.3390/chemistry2020025
Radical-mediated lipid oxidation and the formation of lipid hydroperoxides has been a focal point in the investigation of a number of human pathologies. Lipid peroxidation has long been linked to the inflammatory response and more recently, has been identified as the central tenet of the oxidative cell death mechanism known as ferroptosis. The formation of lipid electrophile-protein adducts has been associated with many of the disorders that involve perturbations of the cellular redox status, but the identities of adducted proteins and the effects of adduction on protein function are mostly unknown. Both cholesterol and 7-dehydrocholesterol (7-DHC), which is the immediate biosynthetic precursor to cholesterol, are oxidizable by species such as ozone and oxygen-centered free radicals. Product mixtures from radical chain processes are particularly complex, with recent studies having expanded the sets of electrophilic compounds formed. Here, we describe recent developments related to the formation of sterol-derived electrophiles and the adduction of these electrophiles to proteins. A framework for understanding sterol peroxidation mechanisms, which has significantly advanced in recent years, as well as the methods for the study of sterol electrophile-protein adduction, are presented in this review.

Chemistry 2020, 2(2), 390-417
https://doi.org/10.3390/chemistry2020025
Radical-mediated lipid oxidation and the formation of lipid hydroperoxides has been a focal point in the investigation of a number of human pathologies. Lipid peroxidation has long been linked to the inflammatory response and more recently, has been identified as the central tenet of the oxidative cell death mechanism known as ferroptosis. The formation of lipid electrophile-protein adducts has been associated with many of the disorders that involve perturbations of the cellular redox status, but the identities of adducted proteins and the effects of adduction on protein function are mostly unknown. Both cholesterol and 7-dehydrocholesterol (7-DHC), which is the immediate biosynthetic precursor to cholesterol, are oxidizable by species such as ozone and oxygen-centered free radicals. Product mixtures from radical chain processes are particularly complex, with recent studies having expanded the sets of electrophilic compounds formed. Here, we describe recent developments related to the formation of sterol-derived electrophiles and the adduction of these electrophiles to proteins. A framework for understanding sterol peroxidation mechanisms, which has significantly advanced in recent years, as well as the methods for the study of sterol electrophile-protein adduction, are presented in this review.

Sunday, May 10, 2020
A novel USP30 inhibitor recapitulates genetic loss of USP30 and sets the trigger for PINK1-PARKIN amplification of mitochondrial ubiquitylation
Emma Rusilowicz-Jones, Jane Jardine, Andreas Kallinos, Adan Pinto-Fernandez, Franziska Guenther, Mariacarmela Giurrandino, Francesco G. Barone, Katy McCarron, Christopher J. Burke, Alejandro Murad, Aitor Martinez, Elena Marcassa, Malte Gersch, Alex Buckmelter, Katherine J. Kayser-Bricker, Frederic Lamoliatte, Akshada Gajbhiye, Simon Davis, Hannah C. Scott, Emma Murphy, Katherine England, Heather Mortiboys, David Komander, Matthias Trost, Benedikt M. Kessler, Stephanos Ioannidis, Michael Ahlijanian, Sylvie Urbé, Michael J. Clague
BioRxiv, 2020
doi: https://doi.org/10.1101/2020.04.16.044206
The mitochondrial deubiquitylase USP30 negatively regulates the selective autophagy of damaged mitochondria. It has been proposed as an actionable target to alleviate the loss of function of the mitophagy pathway governed by the Parkinson’s Disease associated genes PINK1 and PRKN. We present the characterisation of a N-cyano pyrrolidine derived compound, FT3967385, with high selectivity for USP30. The compound is well tolerated with no loss of total mitochondrial mass. We demonstrate that ubiquitylation of TOM20, a component of the outer mitochondrial membrane import machinery that directly interacts with USP30, represents a robust biomarker for both USP30 loss and inhibition. We have conducted proteomics analyses on a SHSY5Y neuroblastoma cell line model to directly compare the effects of genetic loss of USP30 with selective inhibition in an unbiased fashion. We have thereby identified a subset of ubiquitylation events consequent to mitochondrial depolarisation that are USP30 sensitive. Within responsive elements of the ubiquitylome, several components of the outer mitochondrial membrane transport (TOM) complex are most prominent. Thus, our data support a model whereby USP30 can regulate the availability of ubiquitin at the specific site of mitochondrial PINK1 accumulation following membrane depolarisation. In this model, USP30 deubiquitylation of TOM complex components dampens the trigger for the Parkin-dependent amplification of mitochondrial ubiquitylation leading to mitophagy. Accordingly, PINK1 generation of phospho-Ser65 Ubiquitin proceeds more rapidly and to a greater extent in cells either lacking USP30 or subject to USP30 inhibition.

BioRxiv, 2020
doi: https://doi.org/10.1101/2020.04.16.044206
The mitochondrial deubiquitylase USP30 negatively regulates the selective autophagy of damaged mitochondria. It has been proposed as an actionable target to alleviate the loss of function of the mitophagy pathway governed by the Parkinson’s Disease associated genes PINK1 and PRKN. We present the characterisation of a N-cyano pyrrolidine derived compound, FT3967385, with high selectivity for USP30. The compound is well tolerated with no loss of total mitochondrial mass. We demonstrate that ubiquitylation of TOM20, a component of the outer mitochondrial membrane import machinery that directly interacts with USP30, represents a robust biomarker for both USP30 loss and inhibition. We have conducted proteomics analyses on a SHSY5Y neuroblastoma cell line model to directly compare the effects of genetic loss of USP30 with selective inhibition in an unbiased fashion. We have thereby identified a subset of ubiquitylation events consequent to mitochondrial depolarisation that are USP30 sensitive. Within responsive elements of the ubiquitylome, several components of the outer mitochondrial membrane transport (TOM) complex are most prominent. Thus, our data support a model whereby USP30 can regulate the availability of ubiquitin at the specific site of mitochondrial PINK1 accumulation following membrane depolarisation. In this model, USP30 deubiquitylation of TOM complex components dampens the trigger for the Parkin-dependent amplification of mitochondrial ubiquitylation leading to mitophagy. Accordingly, PINK1 generation of phospho-Ser65 Ubiquitin proceeds more rapidly and to a greater extent in cells either lacking USP30 or subject to USP30 inhibition.

Wednesday, May 6, 2020
Efficient targeted degradation via reversible and irreversible covalent PROTACs [@london_lab]
Ronen Gabizon, Amit Shraga, Paul Gehrtz, Ella Livnah, Yamit Shorer, Neta Gurwicz, Liat Avram, Tamar Unger, Hila Aharoni, Shira Albeck, Alexander Brandis, Ziv shulman, Ben-Zion Katz, Yair Herishanu, and Nir London
Journal of the American Chemical Society, 2020
PROteolysis Targeting Chimeras (PROTACs) represent an exciting inhibitory modality with many advantages, including sub-stoichiometric degradation of targets. Their scope, though, is still limited to-date by the requirement for a sufficiently potent target binder. A solution that proved useful in tackling challenging targets is the use of electrophiles to allow irreversible binding to the target. However, such binding will negate the catalytic nature of PROTACs. Reversible covalent PROTACs potentially offer the best of both worlds. They possess the potency and selectivity associated with the formation of the covalent bond, while being able to dissociate and regenerate once the protein target is degraded. Using Bruton’s tyrosine kinase (BTK) as a clinically relevant model system, we show efficient covalent degradation by non-covalent, irreversible covalent and reversible covalent PROTACs, with <10 nM DC50’s and >85% degradation. Our data suggests that part of the degradation by our irreversible covalent PROTACs is driven by reversible binding prior to covalent bond formation, while the reversible covalent PROTACs drive degradation primarily by covalent engagement. The PROTACs showed enhanced inhibition of B cell activation compared to Ibrutinib, and exhibit potent degradation of BTK in patients-derived primary chronic lymphocytic leukemia cells. The most potent reversible covalent PROTAC, RC-3, exhibited enhanced selectivity towards BTK compared to non-covalent and irreversible covalent PROTACs. These compounds may pave the way for the design of covalent PROTACs for a wide variety of challenging targets.
Saturday, May 2, 2020
Discovery of M-808 as a Highly Potent, Covalent, Small-Molecule Inhibitor of the Menin–MLL Interaction with Strong In Vivo Antitumor Activity
Shilin Xu, Angelo Aguilar, Liyue Huang, Tianfeng Xu, Ke Zheng, Donna McEachern, Sally Przybranowski, Caroline Foster, Kaitlin Zawacki, Zhaomin Liu, Krishnapriya Chinnaswamy, Jeanne Stuckey, and Shaomeng Wang
Journal of Medicinal Chemistry, 2020
DOI: 10.1021/acs.jmedchem.0c00547
Targeting the menin–MLL protein–protein interaction is a new therapeutic strategy for the treatment of acute leukemia carrying MLL fusion (MLL leukemia). We describe herein the structure-based optimization of a class of covalent menin inhibitors, which led to the discovery of M-808 (16) as a highly potent and efficacious covalent menin inhibitor. M-808 effectively inhibits leukemia cell growth at low nanomolar concentrations and is capable of achieving partial tumor regression in an MV4;11 xenograft tumor model in mice at a well-tolerated dose schedule. Determination of the co-crystal structure of M-808 in complex with menin provides a structural basis for their high-affinity, covalent interactions. M-808 represents a promising, covalent menin inhibitor for further optimization and evaluation toward developing a new therapy for the treatment of MLL leukemia.

Journal of Medicinal Chemistry, 2020
DOI: 10.1021/acs.jmedchem.0c00547
Targeting the menin–MLL protein–protein interaction is a new therapeutic strategy for the treatment of acute leukemia carrying MLL fusion (MLL leukemia). We describe herein the structure-based optimization of a class of covalent menin inhibitors, which led to the discovery of M-808 (16) as a highly potent and efficacious covalent menin inhibitor. M-808 effectively inhibits leukemia cell growth at low nanomolar concentrations and is capable of achieving partial tumor regression in an MV4;11 xenograft tumor model in mice at a well-tolerated dose schedule. Determination of the co-crystal structure of M-808 in complex with menin provides a structural basis for their high-affinity, covalent interactions. M-808 represents a promising, covalent menin inhibitor for further optimization and evaluation toward developing a new therapy for the treatment of MLL leukemia.

Subscribe to:
Posts (Atom)
-
Design, synthesis and biological evaluation of the activity-based probes for FGFR covalent inhibitorDandan Zhu, Zijian Zheng, Huixin Huang, Xiaojuan Chen, Shuhong Zhang, Zhuchu Chen, Ting Liu, Guangyu Xu, Ying Fu, Yongheng Chen, European Jo...
-
Yoav Shamir, Nir London bioRxiv 2025.03.19.642201 doi: https://doi.org/10.1101/2025.03.19.642201 Recent years have seen an explosion in the...
-
DOI Ansgar Oberheide, Maxime van den Oetelaar, Jakob Scheele, Jan Borggräfe, Semmy Engelen, Michael Sattler, Christian Ottmann, ...
