Fu, L., Jung, Y., Tian, C. et al.
Nat Chem Biol (2023).
https://doi.org/10.1038/s41589-023-01330-5
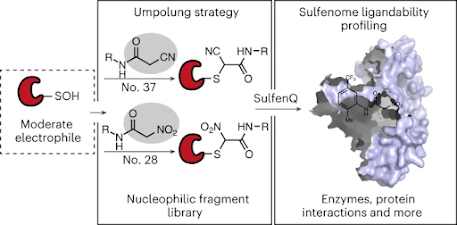
With an eye toward expanding chemistries used for covalent ligand discovery, we elaborated an umpolung strategy that exploits the ‘polarity reversal’ of sulfur when cysteine is oxidized to sulfenic acid, a widespread post-translational modification, for selective bioconjugation with C-nucleophiles. Here we present a global map of a human sulfenome that is susceptible to covalent modification by members of a nucleophilic fragment library. More than 500 liganded sulfenic acids were identified on proteins across diverse functional classes, and, of these, more than 80% were not targeted by electrophilic fragment analogs. We further show that members of our nucleophilic fragment library can impair functional protein–protein interactions involved in nuclear oncoprotein transport and DNA damage repair. Our findings reveal a vast expanse of ligandable sulfenic acids in the human proteome and highlight the utility of nucleophilic small molecules in the fragment-based covalent ligand discovery pipeline, presaging further opportunities using non-traditional chemistries for targeting proteins.