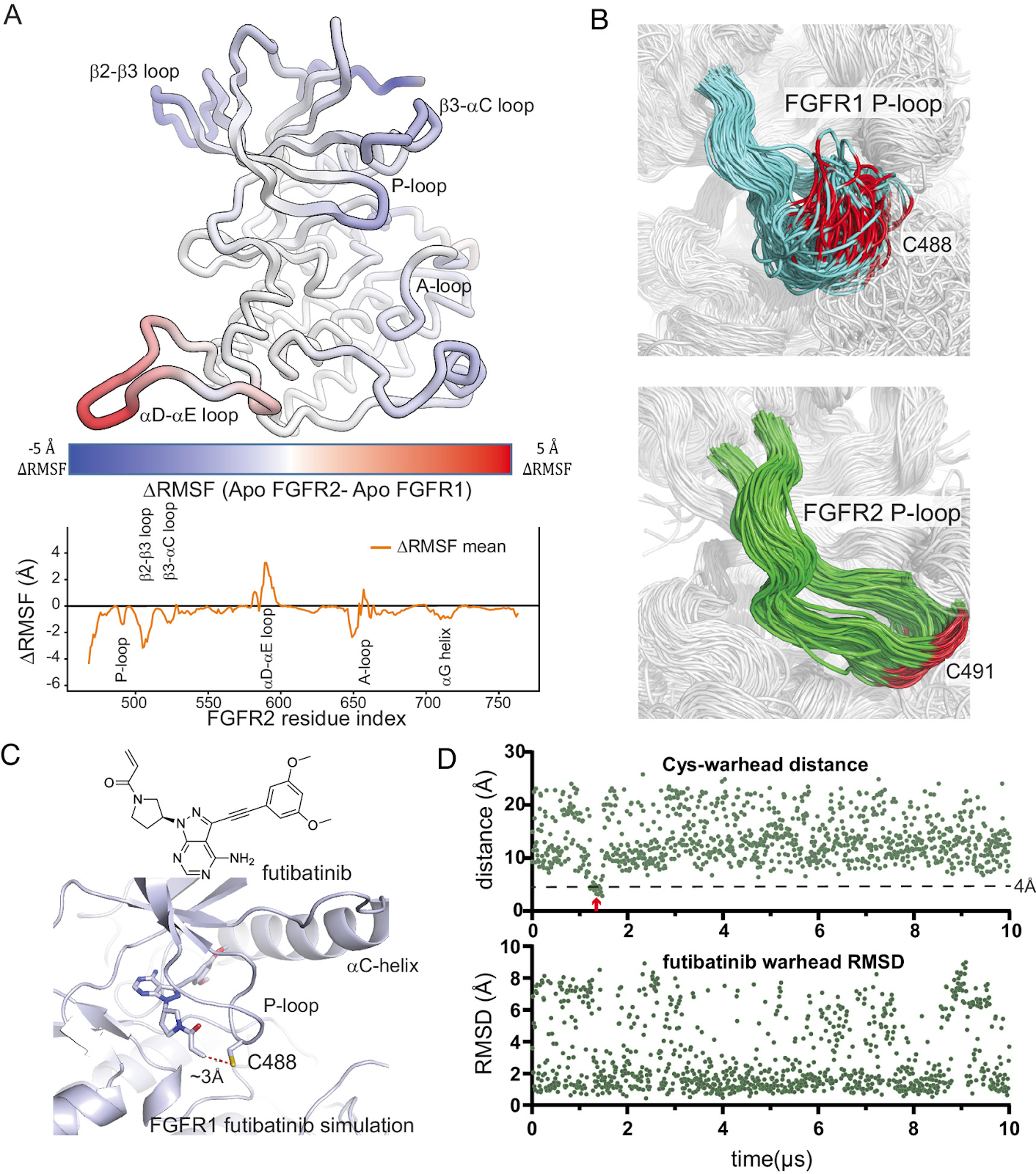
Fibroblast growth factor receptor (FGFR) kinase inhibitors have been shown to be effective in the treatment of intrahepatic cholangiocarcinoma and other advanced solid tumors harboring FGFR2 alterations, but the toxicity of these drugs frequently leads to dose reduction or interruption of treatment such that maximum efficacy cannot be achieved. The most common adverse effects are hyperphosphatemia caused by FGFR1 inhibition and diarrhea due to FGFR4 inhibition, as current therapies are not selective among the FGFRs. Designing selective inhibitors has proved difficult with conventional approaches because the orthosteric sites of FGFR family members are observed to be highly similar in X-ray structures. In this study, aided by analysis of protein dynamics, we designed a selective, covalent FGFR2 inhibitor. In a key initial step, analysis of long-timescale molecular dynamics simulations of the FGFR1 and FGFR2 kinase domains allowed us to identify differential motion in their P-loops, which are located adjacent to the orthosteric site. Using this insight, we were able to design orthosteric binders that selectively and covalently engage the P-loop of FGFR2. Our drug discovery efforts culminated in the development of lirafugratinib (RLY-4008), a covalent inhibitor of FGFR2 that shows substantial selectivity over FGFR1 (~250-fold) and FGFR4 (~5,000-fold) in vitro, causes tumor regression in multiple FGFR2-altered human xenograft models, and was recently demonstrated to be efficacious in the clinic at doses that do not induce clinically significant hyperphosphatemia or diarrhea.