David E. Mortenson, Gabriel J. Brighty, Lars Plate, Grant Bare, Wentao Chen, Suhua Li, Hua Wang, Benjamin F. Cravatt, Stefano Forli , Evan T. Powers, K. Barry Sharpless Ian A. Wilson, and Jeffery W. Kelly
J. Am. Chem. Soc., Article ASAP
DOI: 10.1021/jacs.7b08366
Drug candidates are generally discovered using biochemical screens employing an isolated target protein or by utilizing cell-based phenotypic assays. Both noncovalent and covalent hits emerge from such endeavors. Herein, we exemplify an “Inverse Drug Discovery” strategy in which organic compounds of intermediate complexity harboring weak, but activatable, electrophiles are matched with the protein(s) they react with in cells or cell lysate. An alkyne substructure in each candidate small molecule enables affinity chromatography–mass spectrometry, which produces a list of proteins that each distinct compound reacts with. A notable feature of this approach is that it is agnostic with respect to the cellular proteins targeted. To illustrate this strategy, we employed aryl fluorosulfates, an underexplored class of sulfur(VI) halides, that are generally unreactive unless activated by protein binding. Reversible aryl fluorosulfate binding, correct juxtaposition of protein side chain functional groups, and transition-state stabilization of the S(VI) exchange reaction all seem to be critical for conjugate formation. The aryl fluorosulfates studied thus far exhibit chemoselective reactivity toward Lys and, particularly, Tyr side chains, and can be used to target nonenzymes (e.g., a hormone carrier or a small-molecule carrier protein) as well as enzymes. The “Inverse Drug Discovery” strategy should be particularly attractive as a means to explore latent electrophiles not typically used in medicinal chemistry efforts, until one reacts with a protein target of exceptional interest. Structure–activity data can then be used to enhance the selectivity of conjugate formation or the covalent probe can be used as a competitor to develop noncovalent drug candidates. Here we use the “Inverse Drug Discovery” platform to identify and validate covalent ligands for 11 different human proteins. In the case of one of these proteins, we have identified and validated a small-molecule probe for the first time.
Sunday, December 31, 2017
Saturday, December 16, 2017
Selectively targeting the kinome-conserved lysine of PI3Kδ as a general approach to covalent kinase inhibition
Samuel E Dalton, Lars Dittus, Daniel A. Thomas, Maire A. Convery, Joao Nunes, Jacob T. Bush, John P. Evans, Thilo Werner, Marcus Bantscheff, John A. Murphy, and Sebastien Campos
J. Am. Chem. Soc., 2017
DOI: 10.1021/jacs.7b08979
Selective covalent inhibition of kinases by targeting poorly conserved cysteines has proven highly fruitful to date in the development of chemical probes and approved drugs. However, this approach is limited to ~200 kinases possessing such a cysteine near the ATP-binding pocket. Herein, we report a novel approach to achieve selective, irreversible kinase inhibition, by targeting the conserved catalytic lysine residue. We have illustrated our approach by developing selective, covalent PI3Kδ inhibitors that exhibit nanomolar potency in cellular assays, and a duration of action >48 h in CD4+ T cells. Despite conservation of the lysine residue throughout the kinome, the lead compound shows high levels of selectivity over a selection of lipid and protein kinases in biochemical assays, as well as covalent binding to very few off-target proteins in live-cell proteomic studies. We anticipate this approach could offer an alternative general strategy, to targeting non-conserved cysteines, for the development of selective covalent kinase inhibitors.
J. Am. Chem. Soc., 2017
DOI: 10.1021/jacs.7b08979
Selective covalent inhibition of kinases by targeting poorly conserved cysteines has proven highly fruitful to date in the development of chemical probes and approved drugs. However, this approach is limited to ~200 kinases possessing such a cysteine near the ATP-binding pocket. Herein, we report a novel approach to achieve selective, irreversible kinase inhibition, by targeting the conserved catalytic lysine residue. We have illustrated our approach by developing selective, covalent PI3Kδ inhibitors that exhibit nanomolar potency in cellular assays, and a duration of action >48 h in CD4+ T cells. Despite conservation of the lysine residue throughout the kinome, the lead compound shows high levels of selectivity over a selection of lipid and protein kinases in biochemical assays, as well as covalent binding to very few off-target proteins in live-cell proteomic studies. We anticipate this approach could offer an alternative general strategy, to targeting non-conserved cysteines, for the development of selective covalent kinase inhibitors.
Wednesday, December 6, 2017
Warhead biosynthesis and the origin of structural diversity in hydroxamate metalloproteinase inhibitors
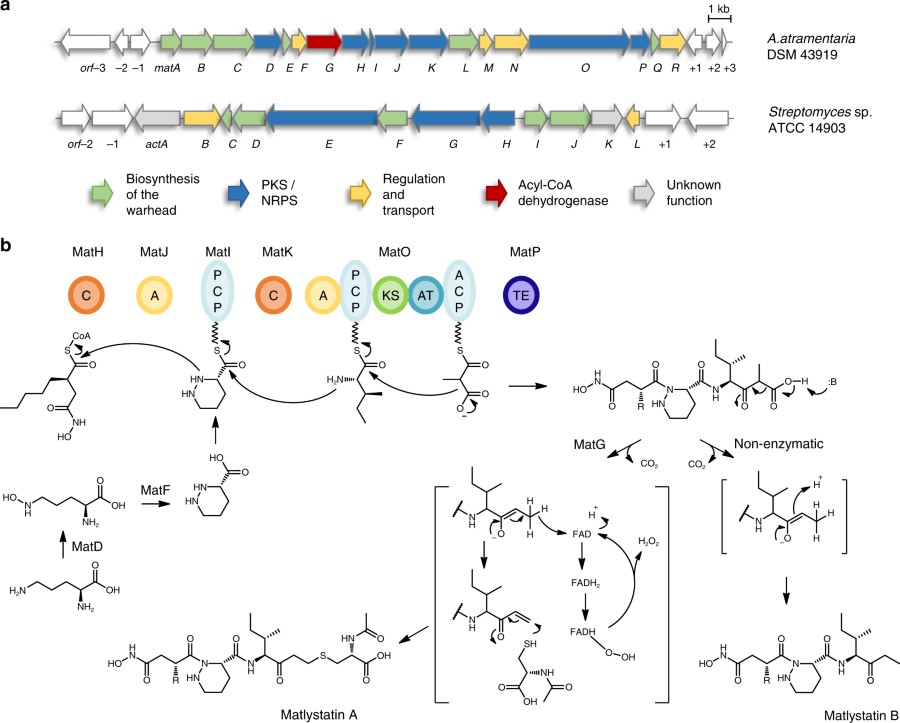
Franziska Leipoldt, Javier Santos-Aberturas, Dennis P. Stegmann, Felix Wolf, Andreas Kulik, Rodney Lacret, Désirée Popadić, Daniela Keinhörster, Norbert Kirchner, Paulina Bekiesch, Harald Gross, Andrew W. Truman & Leonard Kaysser
doi: 10.1038/s41467-017-01975-6
Metalloproteinase inhibitors often feature hydroxamate moieties to facilitate the chelation of metal ions in the catalytic center of target enzymes. Actinonin and matlystatins are potent metalloproteinase inhibitors that comprise rare N-hydroxy-2-pentyl-succinamic acid warheads. Here we report the identification and characterization of their biosynthetic pathways. By gene cluster comparison and a combination of precursor feeding studies, heterologous pathway expression and gene deletion experiments we are able to show that the N-hydroxy-alkyl-succinamic acid warhead is generated by an unprecedented variation of the ethylmalonyl-CoA pathway. Moreover, we present evidence that the remarkable structural diversity of matlystatin congeners originates from the activity of a decarboxylase-dehydrogenase enzyme with high similarity to enzymes that form epoxyketones. We further exploit this mechanism to direct the biosynthesis of non-natural matlystatin derivatives. Our work paves the way for follow-up studies on these fascinating pathways and allows the identification of new protease inhibitors by genome mining.
Tuesday, December 5, 2017
Chemoselective Installation of Amine Bonds on Proteins through Aza-Michael Ligation
Allyson M. Freedy, Maria J. Matos, Omar Boutureira, Francisco Corzana, Ana Guerreiro, Padma Akkapeddi, Víctor J. Somovilla, Tiago Rodrigues, Karl Nicholls, Bangwen Xie, Gonzalo Jiménez-Osés, Kevin M. Brindle, André A. Neves, & Gonçalo J. L. Bernardes
J. Am. Chem. Soc., 2017 ASAP
DOI: 10.1021/jacs.7b10702
Chemical modification of proteins is essential for a variety of important diagnostic and therapeutic applications. Many strategies developed to date lack chemo- and regioselectivity as well as result in non-native linkages that may suffer from instability in vivo and adversely affect the protein’s structure and function. We describe here the reaction of N-nucleophiles with the amino acid dehydroalanine (Dha) in a protein context. When Dha is chemically installed in proteins, the addition of a wide-range N-nucleophiles enables the rapid formation of amine linkages (secondary and tertiary) in a chemoselective manner under mild, biocompatible conditions. These new linkages are stable at a wide range of pH values (pH 2.8 to 12.8), under reducing conditions (biological thiols such as glutathione) and in human plasma. This method is demonstrated for three proteins and is shown to be fully compatible with disulfide bridges, as evidenced by the selective modification of recombinant albumin that displays 17 structurally relevant disulfides. The practicability and utility of our approach is further demonstrated by the construction of a chemically modified C2A domain of Synaptotagmin-I protein that retains its ability to preferentially bind to apoptotic cells at a level comparable to the native protein. Importantly, the method was useful for building a homogeneous antibody-drug conjugate with a precise drug-to-antibody ratio of 2. The kinase inhibitor crizotinib was directly conjugated to Dha through its piperidine motif, and its antibody-mediated intracellular delivery results in 10-fold improvement of its cancer cell-killing efficacy. The simplicity and exquisite site-selectivity of the aza-Michael ligation described herein allows the construction of stable secondary and tertiary amine-linked protein conjugates without affecting the structure and function of biologically relevant proteins.

Friday, November 17, 2017
Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products
Elizabeth A. Grossman, Carl C. Ward, Jessica N. Spradlin, Leslie A. Bateman, Tucker R. Huffman, David K. Miyamoto, Jordan I. Kleinman, Daniel K. Nomura
Cell Chemical Biology, 2017
doi: 10.1016/j.chembiol.2017.08.013
Many natural products that show therapeutic activities are often difficult to synthesize or isolate and have unknown targets, hindering their development as drugs. Identifying druggable hotspots targeted by covalently acting anti-cancer natural products can enable pharmacological interrogation of these sites with more synthetically tractable compounds. Here, we used chemoproteomic platforms to discover that the anti-cancer natural product withaferin A targets C377 on the regulatory subunit PPP2R1A of the tumor-suppressor protein phosphatase 2A (PP2A) complex leading to activation of PP2A activity, inactivation of AKT, and impaired breast cancer cell proliferation. We developed a more synthetically tractable cysteine-reactive covalent ligand, JNS 1-40, that selectively targets C377 of PPP2R1A to impair breast cancer signaling, proliferation, and in vivo tumor growth. Our study highlights the utility of using chemoproteomics to map druggable hotspots targeted by complex natural products and subsequently interrogating these sites with more synthetically tractable covalent ligands for cancer therapy.

Cell Chemical Biology, 2017
doi: 10.1016/j.chembiol.2017.08.013
Many natural products that show therapeutic activities are often difficult to synthesize or isolate and have unknown targets, hindering their development as drugs. Identifying druggable hotspots targeted by covalently acting anti-cancer natural products can enable pharmacological interrogation of these sites with more synthetically tractable compounds. Here, we used chemoproteomic platforms to discover that the anti-cancer natural product withaferin A targets C377 on the regulatory subunit PPP2R1A of the tumor-suppressor protein phosphatase 2A (PP2A) complex leading to activation of PP2A activity, inactivation of AKT, and impaired breast cancer cell proliferation. We developed a more synthetically tractable cysteine-reactive covalent ligand, JNS 1-40, that selectively targets C377 of PPP2R1A to impair breast cancer signaling, proliferation, and in vivo tumor growth. Our study highlights the utility of using chemoproteomics to map druggable hotspots targeted by complex natural products and subsequently interrogating these sites with more synthetically tractable covalent ligands for cancer therapy.
Tuesday, November 14, 2017
Can Relative Binding Free Energy Predict Selectivity of Reversible Covalent Inhibitors?
Payal Chatterjee, Wesley M. Botello-Smith, Han Zhang, Li Qian, Abdelaziz Alsamarah, David Kent, Jerome J. Lacroix, Michel Baudry, and Yun Luo
J. Am. Chem. Soc., 2017
doi: 10.1021/jacs.7b08938
Reversible covalent inhibitors have many clinical advantages over noncovalent or covalent drugs. However, apart from selecting a warhead, substantial efforts in design and synthesis are needed to optimize noncovalent interactions to improve target-selective binding. Computational prediction of binding affinity for reversible covalent inhibitors presents a unique challenge since the binding process consists of multiple steps, which are not necessarily independent of each other. In this study, we lay out the relation between relative binding free energy and the overall reversible covalent binding affinity using a two-state binding model. To prove the concept, we employed free energy perturbation (FEP) coupled with λ-exchange molecular dynamics method to calculate the binding free energy of a series of α-ketoamide analogs relative to a common warhead scaffold, in both noncovalent and covalent bond states, and for two highly homologous proteases, calpain-1 and calpain-2. We conclude that covalent binding affinity alone, in general, can be used to predict reversible covalent binding selectivity. However, exceptions may exist. Therefore, we also discuss the conditions under which the noncovalent binding step is no longer negligible and propose a novel approach that combines the relative FEP calculations with a single QM/MM calculation of warhead to predict the binding affinity and binding kinetics for a large number of reversible covalent inhibitors. Our FEP calculations also revealed that covalent and noncovalent states of an inhibitor do not necessarily exhibit the same selectivity. Thus, investigating both binding states, as well as the kinetics will provide extremely useful information for optimizing reversible covalent inhibitors.
J. Am. Chem. Soc., 2017
doi: 10.1021/jacs.7b08938
Reversible covalent inhibitors have many clinical advantages over noncovalent or covalent drugs. However, apart from selecting a warhead, substantial efforts in design and synthesis are needed to optimize noncovalent interactions to improve target-selective binding. Computational prediction of binding affinity for reversible covalent inhibitors presents a unique challenge since the binding process consists of multiple steps, which are not necessarily independent of each other. In this study, we lay out the relation between relative binding free energy and the overall reversible covalent binding affinity using a two-state binding model. To prove the concept, we employed free energy perturbation (FEP) coupled with λ-exchange molecular dynamics method to calculate the binding free energy of a series of α-ketoamide analogs relative to a common warhead scaffold, in both noncovalent and covalent bond states, and for two highly homologous proteases, calpain-1 and calpain-2. We conclude that covalent binding affinity alone, in general, can be used to predict reversible covalent binding selectivity. However, exceptions may exist. Therefore, we also discuss the conditions under which the noncovalent binding step is no longer negligible and propose a novel approach that combines the relative FEP calculations with a single QM/MM calculation of warhead to predict the binding affinity and binding kinetics for a large number of reversible covalent inhibitors. Our FEP calculations also revealed that covalent and noncovalent states of an inhibitor do not necessarily exhibit the same selectivity. Thus, investigating both binding states, as well as the kinetics will provide extremely useful information for optimizing reversible covalent inhibitors.
Sunday, November 5, 2017
Development of 5N-Bicalutamide, a High-Affinity Reversible Covalent Antiandrogen
Felipe de Jesus Cortez, Phuong Nguyen, Charles Truillet, Boxue Tian, Kristopher M. Kuchenbecker, Michael J. Evans, Paul Webb, Matthew P. Jacobson , Robert J. Fletterick, and Pamela M. England
ACS Chem. Biol., 2017
DOI: 10.1021/acschembio.7b00702

Tuesday, October 31, 2017
Proteome-wide Map of Targets of T790M-EGFR-Directed Covalent Inhibitors
Sherry Niessen,, Melissa M. Dix, Sabrina Barbas, Zachary E. Potter, Shuyan Lu, Oleg Brodsky, Simon Planken, Douglas Behenna, Chau Almaden, Ketan S. Gajiwala, Kevin Ryan, RoseAnn Ferre, Michael R. Lazear, Matthew M. Hayward, John C. Kath, Benjamin F. Cravatt
Cell Chemical Biology, 2017 doi: 10.1016/j.chembiol.2017.08.017
Cell Chemical Biology, 2017 doi: 10.1016/j.chembiol.2017.08.017
Abstract
Patients with non-small cell lung cancers that have kinase-activating epidermal growth factor receptor (EGFR) mutations are highly responsive to first- and second-generation EGFR inhibitors. However, these patients often relapse due to a secondary, drug-resistant mutation in EGFR whereby the gatekeeper threonine is converted to methionine (T790M). Several third-generation EGFR inhibitors have been developed that irreversibly inactivate T790M-EGFR while sparing wild-type EGFR, thus reducing epithelium-based toxicities. Using chemical proteomics, we show here that individual T790M-EGFR inhibitors exhibit strikingly distinct off-target profiles in human cells. The FDA-approved drug osimertinib (AZD9291), in particular, was found to covalently modify cathepsins in cell and animal models, which correlated with lysosomal accumulation of the drug. Our findings thus show how chemical proteomics can be used to differentiate covalent kinase inhibitors based on global selectivity profiles in living systems and identify specific off-targets of these inhibitors that may affect drug activity and safety.

Monday, October 30, 2017
Sequence-Based Prediction of Cysteine Reactivity Using Machine Learning
Haobo Wang, Xuemin Chen, Can Li, Yuan Liu, Fan Yang, and Chu Wang
Biochemistry, 2017 DOI: 10.1021/acs.biochem.7b00897
As one of the most intrinsically reactive amino acids, cysteine carries a variety of important biochemical functions, including catalysis and redox regulation. Discovery and characterization of cysteines with heightened reactivity will help annotate protein functions. Chemical proteomic methods have been used to quantitatively profile cysteine reactivity in native proteomes, showing a strong correlation between the chemical reactivity of a cysteine and its functionality; however, the relationship between the cysteine reactivity and its local sequence has not yet been systematically explored. Herein, we report a machine learning method, sbPCR (sequence-based prediction of cysteine reactivity), which combines the basic local alignment search tool, truncated composition of k-spaced amino acid pair analysis, and support vector machine to predict cysteines with hyper-reactivity based on only local sequence features. Using a benchmark set compiled from hyper-reactive cysteines in human proteomes, our method can achieve a prediction accuracy of 98%, a precision of 95%, and a recall ratio of 89%. We utilized these governing features of local sequence motifs to expand the prediction to potential hyper-reactive cysteines in other proteomes deposited in the UniProt database. We validated our predictions in Escherichia coli by activity-based protein profiling and discovered a hyper-reactive cysteine from a functionally uncharacterized protein, YecH. Biochemical analysis suggests that the hyper-reactive cysteine might be involved in metal binding. Our computational method provides a large inventory of potential hyper-reactive cysteines in proteomes and is highly complementary to other experimental approaches to guide systematic annotation of protein functions in the postgenome era.

Biochemistry, 2017 DOI: 10.1021/acs.biochem.7b00897

Thursday, October 12, 2017
Targeting the Protein Kinases Cysteinome
Apirat Chaikuad, Pierre Koch, Stefan Laufer, and Stefan Knapp
Angew. Chem. Int. Ed. 2017
doi: 10.1002/anie.201707875
Drugs that function by covalent bond formation represent a considerable fraction of our repository of effective medicines but safety concerns and the complexity of developing covalent inhibitors has rendered covalent targeting a less attractive strategy for rational drug design. The recent approval of four covalent kinase inhibitors and the development of highly potent covalent kinase probes with exceptional selectivity has raised significant interest in industry and academic research and validated the concept of covalent kinase targeting for clinical applications. The abundance of cysteines at diverse positions in and around the kinase active site suggests that a large fraction of kinases can be targeted by covalent inhibitors. Here we review recent developments of this rapidly growing area in kinase drug development and highlight the unique opportunities and challenges of this strategy.
Angew. Chem. Int. Ed. 2017
doi: 10.1002/anie.201707875
Drugs that function by covalent bond formation represent a considerable fraction of our repository of effective medicines but safety concerns and the complexity of developing covalent inhibitors has rendered covalent targeting a less attractive strategy for rational drug design. The recent approval of four covalent kinase inhibitors and the development of highly potent covalent kinase probes with exceptional selectivity has raised significant interest in industry and academic research and validated the concept of covalent kinase targeting for clinical applications. The abundance of cysteines at diverse positions in and around the kinase active site suggests that a large fraction of kinases can be targeted by covalent inhibitors. Here we review recent developments of this rapidly growing area in kinase drug development and highlight the unique opportunities and challenges of this strategy.
Wednesday, October 11, 2017
Covalent lectin inhibition and application in bacterial biofilm imaging
Stefanie Wagner, Dirk Hauck, Michael Hoffmann, Roman Sommer, Ines Joachim, Rolf Müller, Anne Imberty, Annabelle Varrot, Alexander Titz
Angew. Chem. 2017
DOI: 10.1002/ange.201709368
Biofilm formation by pathogenic bacteria is a hallmark of chronic infections. In many cases, lectins play key roles in establishing biofilms. The pathogen Pseudomonas aeruginosa often exhibiting various drug resistances employs its lectins LecA and LecB as virulence factors and biofilm building blocks. Therefore, inhibition of the function of these proteins is thought to have potential in developing 'pathoblockers' preventing biofilm formation and virulence. Here, we describe for the first time a covalent lectin inhibitor specific to a carbohydrate binding site. In addition we report its application in the LecA-specific in vitro imaging of biofilms formed by P. aeruginosa.
Angew. Chem. 2017
DOI: 10.1002/ange.201709368
Biofilm formation by pathogenic bacteria is a hallmark of chronic infections. In many cases, lectins play key roles in establishing biofilms. The pathogen Pseudomonas aeruginosa often exhibiting various drug resistances employs its lectins LecA and LecB as virulence factors and biofilm building blocks. Therefore, inhibition of the function of these proteins is thought to have potential in developing 'pathoblockers' preventing biofilm formation and virulence. Here, we describe for the first time a covalent lectin inhibitor specific to a carbohydrate binding site. In addition we report its application in the LecA-specific in vitro imaging of biofilms formed by P. aeruginosa.
Tuesday, October 10, 2017
Cytosolic Delivery of Proteins by Bioreversible Esterification
Kalie A. Mix, Jo E. Lomax, and Ronald T. Raines
J. Am. Chem. Soc., Article ASAP
DOI: 10.1021/jacs.7b06597
Cloaking its carboxyl groups with a hydrophobic moiety is shown to enable a protein to enter the cytosol of a mammalian cell. Diazo compounds derived from (p-methylphenyl)glycine were screened for the ability to esterify the green fluorescent protein (GFP) in an aqueous environment. Esterification of GFP with 2-diazo-2-(p-methylphenyl)-N,N-dimethylacetamide was efficient. The esterified protein entered the cytosol by traversing the plasma membrane directly, like a small-molecule prodrug. As with prodrugs, the nascent esters are substrates for endogenous esterases, which regenerate native protein. Thus, esterification could provide a general means to deliver native proteins to the cytosol.

J. Am. Chem. Soc., Article ASAP
DOI: 10.1021/jacs.7b06597
Cloaking its carboxyl groups with a hydrophobic moiety is shown to enable a protein to enter the cytosol of a mammalian cell. Diazo compounds derived from (p-methylphenyl)glycine were screened for the ability to esterify the green fluorescent protein (GFP) in an aqueous environment. Esterification of GFP with 2-diazo-2-(p-methylphenyl)-N,N-dimethylacetamide was efficient. The esterified protein entered the cytosol by traversing the plasma membrane directly, like a small-molecule prodrug. As with prodrugs, the nascent esters are substrates for endogenous esterases, which regenerate native protein. Thus, esterification could provide a general means to deliver native proteins to the cytosol.
Sunday, October 8, 2017
Proteome-wide Map of Targets of T790M-EGFR-Directed Covalent Inhibitors
Sherry Niessen, Melissa M.Dix, Sabrina Barbas, Zachary Potter. Shuyan Lu, Oleg Brodsky, Simon Planken, Douglas Behenna, Chau Almaden, Ketan S. Gajiwala, Kevin Ryan, Rose Ann Ferre, Michael R. Lazear. Matthew M.Hayward, John C. Kath, Benjamin F. Cravatt
Cell Chemical Biology, 2017
doi: 10.1016/j.chembiol.2017.08.017
Patients with non-small cell lung cancers that have kinase-activating epidermal growth factor receptor (EGFR) mutations are highly responsive to first- and second-generation EGFR inhibitors. However, these patients often relapse due to a secondary, drug-resistant mutation in EGFR whereby the gatekeeper threonine is converted to methionine (T790M). Several third-generation EGFR inhibitors have been developed that irreversibly inactivate T790M-EGFR while sparing wild-type EGFR, thus reducing epithelium-based toxicities. Using chemical proteomics, we show here that individual T790M-EGFR inhibitors exhibit strikingly distinct off-target profiles in human cells. The FDA-approved drug osimertinib (AZD9291), in particular, was found to covalently modify cathepsins in cell and animal models, which correlated with lysosomal accumulation of the drug. Our findings thus show how chemical proteomics can be used to differentiate covalent kinase inhibitors based on global selectivity profiles in living systems and identify specific off-targets of these inhibitors that may affect drug activity and safety.

Cell Chemical Biology, 2017
doi: 10.1016/j.chembiol.2017.08.017
Patients with non-small cell lung cancers that have kinase-activating epidermal growth factor receptor (EGFR) mutations are highly responsive to first- and second-generation EGFR inhibitors. However, these patients often relapse due to a secondary, drug-resistant mutation in EGFR whereby the gatekeeper threonine is converted to methionine (T790M). Several third-generation EGFR inhibitors have been developed that irreversibly inactivate T790M-EGFR while sparing wild-type EGFR, thus reducing epithelium-based toxicities. Using chemical proteomics, we show here that individual T790M-EGFR inhibitors exhibit strikingly distinct off-target profiles in human cells. The FDA-approved drug osimertinib (AZD9291), in particular, was found to covalently modify cathepsins in cell and animal models, which correlated with lysosomal accumulation of the drug. Our findings thus show how chemical proteomics can be used to differentiate covalent kinase inhibitors based on global selectivity profiles in living systems and identify specific off-targets of these inhibitors that may affect drug activity and safety.

Wednesday, September 27, 2017
Covalent binding design strategy: A prospective method for discovery of potent targeted anticancer agents
Luhong Wang, Jingyuan Zhao, Yao Yao, Changyuan Wang, Jianbin Zhang, Xiaohong Shu, Xiuli Sun, Yanxia Li, Kexin Liu, Hong Yuan, Xiaodong Ma
European Journal of Medicinal Chemistry, 2017
DOI: 10.1016/j.ejmech.2017.09.024
Cancer remains the most serious disease that threatens human health. Molecularly targeted cancer therapies, specifically small-molecule protein kinase inhibitors, form an important part of cancer therapy. Targeted covalent modification represents a proven approach to drug discovery with the recent FDA approvals of afatanib, ibrutinib, and osimertinib agents, which were designed to undergo an irreversible hetero-Michael addition reaction with a unique cysteine residue of a specific protein. Covalent inhibitors possess numerous advantages, including increased biochemical efficacy, longer duration of action, the high potential for improved therapeutic index due to lower effective dose, and the potential to inhibit certain drug resistance mechanisms. In this regard, the novel targeted anticancer agents whose activity is presumably dependent upon a hetero-Michael addition reaction with thiols are summarized in this article.
European Journal of Medicinal Chemistry, 2017
DOI: 10.1016/j.ejmech.2017.09.024
Cancer remains the most serious disease that threatens human health. Molecularly targeted cancer therapies, specifically small-molecule protein kinase inhibitors, form an important part of cancer therapy. Targeted covalent modification represents a proven approach to drug discovery with the recent FDA approvals of afatanib, ibrutinib, and osimertinib agents, which were designed to undergo an irreversible hetero-Michael addition reaction with a unique cysteine residue of a specific protein. Covalent inhibitors possess numerous advantages, including increased biochemical efficacy, longer duration of action, the high potential for improved therapeutic index due to lower effective dose, and the potential to inhibit certain drug resistance mechanisms. In this regard, the novel targeted anticancer agents whose activity is presumably dependent upon a hetero-Michael addition reaction with thiols are summarized in this article.
Tuesday, September 26, 2017
Structure–Activity Relationships of Potent, Targeted Covalent Inhibitors That Abolish Both the Transamidation and GTP Binding Activities of Human Tissue Transglutaminase
Abdullah Akbar, Nicole M. R. McNeil, Marie R. Albert, Viviane Ta, Gautam Adhikary, Karine Bourgeois, Richard L. Eckert, and Jeffrey W. Keillor
J. Med. Chem., Article ASAP

Human tissue transglutaminase (hTG2) is a multifunctional enzyme. It is primarily known for its calcium-dependent transamidation activity that leads to formation of an isopeptide bond between glutamine and lysine residues found on the surface of proteins, but it is also a GTP binding protein. Overexpression and unregulated hTG2 activity have been associated with numerous human diseases, including cancer stem cell survival and metastatic phenotype. Herein, we present a series of targeted covalent inhibitors (TCIs) based on our previously reported Cbz-Lys scaffold. From this structure–activity relationship (SAR) study, novel irreversible inhibitors were identified that block the transamidation activity of hTG2 and allosterically abolish its GTP binding ability with a high degree of selectivity and efficiency (kinact/KI > 105 M–1 min–1). One optimized inhibitor (VA4) was also shown to inhibit epidermal cancer stem cell invasion with an EC50 of 3.9 μM, representing a significant improvement over our previously reported “hit” NC9.
Impact of the structures of macrocyclic Michael acceptors on covalent proteasome inhibition
S. Kitahata,a F. Yakushijiab and S. Ichikawa
Chem. Sci., 2017, 8, 6959-6963
Molecules that have a reactive functional group within a macrocycle represent a class of covalent inhibitor. The relationship between reactivity and affinity for the target is cooperative and complicated. An understanding and characterization of this class of inhibitor are vital for the development of covalent inhibitors as drug candidates. Herein, we describe a systematic analysis of structure–activity relationships using a series of syringolin analogues, which are irreversible covalent inhibitors of proteasomes. We investigate the detailed mechanistic effects of the macrocycles on affinity and reaction rate.

Chem. Sci., 2017, 8, 6959-6963
DOI: 10.1039/C7SC02941A
Tuesday, September 19, 2017
Differential Kinobeads Profiling for Target Identification of Irreversible Kinase Inhibitors
Lars Dittus, Thilo Werner, Marcel Muelbaier and Marcus Bantscheff
GlaxoSmithKline, Meyerhofstrasse 1, D-69117 Heidelberg, Germany
Chemoproteomics profiling of kinase inhibitors with kinobeads enables the assessment of inhibitor potency and selectivity for endogenously expressed protein kinases in cell lines and tissues. Using a small panel of targeted covalent inhibitors, we demonstrate the importance of measuring covalent target binding in live cells. We present a differential kinobeads profiling strategy for covalent kinase inhibitors where a compound is added either to live cells or to a cell extract that enables the comprehensive assessment of inhibitor selectivity for covalent and noncovalent targets. We found that Acalabrutinib, CC-292, and Ibrutinib potently and covalently bind TEC family kinases, but only Ibrutinib also potently binds to BLK. ZAK was identified as a submicromolar affinity Ibrutinib off-target due to covalent modification of Cys22. In contrast to Ibrutinib, 5Z-7-Oxozeaenol reacted with Cys150 next to the DFG loop, demonstrating an alternative route to covalent inactivation of this kinase, e.g., to inhibit canonical TGF-β dependent processes.
GlaxoSmithKline, Meyerhofstrasse 1, D-69117 Heidelberg, Germany
ACS Chem. Biol., Article ASAP
Chemoproteomics profiling of kinase inhibitors with kinobeads enables the assessment of inhibitor potency and selectivity for endogenously expressed protein kinases in cell lines and tissues. Using a small panel of targeted covalent inhibitors, we demonstrate the importance of measuring covalent target binding in live cells. We present a differential kinobeads profiling strategy for covalent kinase inhibitors where a compound is added either to live cells or to a cell extract that enables the comprehensive assessment of inhibitor selectivity for covalent and noncovalent targets. We found that Acalabrutinib, CC-292, and Ibrutinib potently and covalently bind TEC family kinases, but only Ibrutinib also potently binds to BLK. ZAK was identified as a submicromolar affinity Ibrutinib off-target due to covalent modification of Cys22. In contrast to Ibrutinib, 5Z-7-Oxozeaenol reacted with Cys150 next to the DFG loop, demonstrating an alternative route to covalent inactivation of this kinase, e.g., to inhibit canonical TGF-β dependent processes.
Monday, September 18, 2017
Quantification and Theoretical Analysis of the Electrophilicities of Michael Acceptors
Dominik S. Allgäuer, Harish Jangra, Haruyasu Asahara , Zhen Li, Quan Chen, Hendrik Zipse, Armin R. Ofial, and Herbert Mayr
J. Am. Chem. Soc., Article ASAP
DOI: 10.1021/jacs.7b05106
In order to quantify the electrophilic reactivities of common Michael acceptors, we measured the kinetics of the reactions of monoacceptor-substituted ethylenes (H2C═CH-Acc, 1) and styrenes (PhCH═CH-Acc, 2) with pyridinium ylides 3, sulfonium ylide 4, and sulfonyl-substituted chloromethyl anion 5. Substitution of the 57 measured second-order rate constants (log k) and the previously reported nucleophile-specific parameters N and sN for 3–5 into the correlation log k = sN(E + N) allowed us to calculate 15 new empirical electrophilicity parameters E for Michael acceptors 1 and 2. The use of the same parameters sN, N, and E for these different types of reactions shows that all reactions proceed via a common rate-determining step, the nucleophilic attack of 3–5 at the Michael acceptors with formation of acyclic intermediates, which subsequently cyclize to give tetrahydroindolizines (stepwise 1,3-dipolar cycloadditions with 3) and cyclopropanes (with 4 and 5), respectively. The electrophilicity parameters E thus determined can be used to calculate the rates of the reactions of Michael acceptors 1 and 2 with any nucleophile of known N and sN. DFT calculations were performed to confirm the suggested reaction mechanisms and to elucidate the origin of the electrophilic reactivities. While electrophilicities E correlate poorly with the LUMO energies and with Parr’s electrophilicity index ω, good correlations were found between the experimentally observed electrophilic reactivities of 44 Michael acceptors and their calculated methyl anion affinities, particularly when solvation by dimethyl sulfoxide was taken into account by applying the SMD continuum solvation model. Because of the large structural variety of Michael acceptors considered for these correlations, which cover a reactivity range of 17 orders of magnitude, we consider the calculation of methyl anion affinities to be the method of choice for a rapid estimate of electrophilic reactivities.
J. Am. Chem. Soc., Article ASAP
DOI: 10.1021/jacs.7b05106
In order to quantify the electrophilic reactivities of common Michael acceptors, we measured the kinetics of the reactions of monoacceptor-substituted ethylenes (H2C═CH-Acc, 1) and styrenes (PhCH═CH-Acc, 2) with pyridinium ylides 3, sulfonium ylide 4, and sulfonyl-substituted chloromethyl anion 5. Substitution of the 57 measured second-order rate constants (log k) and the previously reported nucleophile-specific parameters N and sN for 3–5 into the correlation log k = sN(E + N) allowed us to calculate 15 new empirical electrophilicity parameters E for Michael acceptors 1 and 2. The use of the same parameters sN, N, and E for these different types of reactions shows that all reactions proceed via a common rate-determining step, the nucleophilic attack of 3–5 at the Michael acceptors with formation of acyclic intermediates, which subsequently cyclize to give tetrahydroindolizines (stepwise 1,3-dipolar cycloadditions with 3) and cyclopropanes (with 4 and 5), respectively. The electrophilicity parameters E thus determined can be used to calculate the rates of the reactions of Michael acceptors 1 and 2 with any nucleophile of known N and sN. DFT calculations were performed to confirm the suggested reaction mechanisms and to elucidate the origin of the electrophilic reactivities. While electrophilicities E correlate poorly with the LUMO energies and with Parr’s electrophilicity index ω, good correlations were found between the experimentally observed electrophilic reactivities of 44 Michael acceptors and their calculated methyl anion affinities, particularly when solvation by dimethyl sulfoxide was taken into account by applying the SMD continuum solvation model. Because of the large structural variety of Michael acceptors considered for these correlations, which cover a reactivity range of 17 orders of magnitude, we consider the calculation of methyl anion affinities to be the method of choice for a rapid estimate of electrophilic reactivities.
Tuesday, September 5, 2017
Structure-Activity Relationships of potent, targeted covalent inhibitors that abolish both the transamidation and GTP binding activities of human tissue transglutaminase
Abdullah Akbar, Nicole M.R. McNeil, Marie R. Albert, Viviane Ta, Gautam Adhikary, Karine Bourgeois, Richard L. Eckert, and Jeffrey W. Keillor
J. Med. Chem., 2017
DOI: 10.1021/acs.jmedchem.7b01070
Human tissue transglutaminase (hTG2) is a multifunctional enzyme. It is primarily known for its calcium-dependent transamidation activity that leads to formation of an isopeptide bond between glutamine and lysine residues found on the surface of proteins, but it is also a GTP binding protein. Overexpression and unregulated hTG2 activity has been associated with numerous human diseases, including cancer stem cell survival and metastatic phenotype. Herein, we present a series of targeted covalent inhibitors (TCIs) based on our previously reported Cbz-Lys scaffold. From this structure-activity relationship (SAR) study, novel irreversible inhibitors were identified that block the transamidation activity of hTG2 and allosterically abolish its GTP binding ability with a high degree of selectivity and efficiency (kinact/KI > 105 M-1min-1). One optimized inhibitor (VA4) was also shown to inhibit epidermal cancer stem cell invasion with an EC50 of 3.9 µM, representing a significant improvement over our previously reported ‘hit’ NC9.
J. Med. Chem., 2017
DOI: 10.1021/acs.jmedchem.7b01070
Human tissue transglutaminase (hTG2) is a multifunctional enzyme. It is primarily known for its calcium-dependent transamidation activity that leads to formation of an isopeptide bond between glutamine and lysine residues found on the surface of proteins, but it is also a GTP binding protein. Overexpression and unregulated hTG2 activity has been associated with numerous human diseases, including cancer stem cell survival and metastatic phenotype. Herein, we present a series of targeted covalent inhibitors (TCIs) based on our previously reported Cbz-Lys scaffold. From this structure-activity relationship (SAR) study, novel irreversible inhibitors were identified that block the transamidation activity of hTG2 and allosterically abolish its GTP binding ability with a high degree of selectivity and efficiency (kinact/KI > 105 M-1min-1). One optimized inhibitor (VA4) was also shown to inhibit epidermal cancer stem cell invasion with an EC50 of 3.9 µM, representing a significant improvement over our previously reported ‘hit’ NC9.
Saturday, September 2, 2017
Lysine-Targeting Covalent Inhibitors
Jonathan Pettinger, Keith Jones, Matthew David Cheeseman
Angewandte Chemie International Edition, 2017
DOI: 10.1002/anie.201707630
Targeted covalent inhibitors have gained widespread attention in drug discovery as a validated method to circumvent acquired resistance in oncology. This strategy exploits small molecule/protein crystal structures to design tight-binding ligands with appropriately positioned electrophilic warheads. Whilst most focus has been on targeting binding site cysteine residues, targeting nucleophilic lysine residues can also represent a viable approach to irreversible inhibition. However, owing to the basicity of the ε-amino group in lysine, this strategy generates a number of specific challenges. Herein, we review the key principles for inhibitor design, give historical examples and present recent developments that demonstrate its potential for future drug discovery.
Angewandte Chemie International Edition, 2017
DOI: 10.1002/anie.201707630
Targeted covalent inhibitors have gained widespread attention in drug discovery as a validated method to circumvent acquired resistance in oncology. This strategy exploits small molecule/protein crystal structures to design tight-binding ligands with appropriately positioned electrophilic warheads. Whilst most focus has been on targeting binding site cysteine residues, targeting nucleophilic lysine residues can also represent a viable approach to irreversible inhibition. However, owing to the basicity of the ε-amino group in lysine, this strategy generates a number of specific challenges. Herein, we review the key principles for inhibitor design, give historical examples and present recent developments that demonstrate its potential for future drug discovery.
Monday, August 21, 2017
Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: From design to protein crystallography
Albert H. Chan, Won-Gil Lee, Krasimir A. Spasov, José A. Cisneros, Shalley N. Kudalkar, Zaritza O. Petrova, Amanda B. Buckingham, Karen S. Anderson, and William L. Jorgensen
PNAS 2017
doi: 10.1073/pnas.1711463114
Development of resistance remains a major challenge for drugs to treat HIV-1 infections, including those targeting the essential viral polymerase, HIV-1 reverse transcriptase (RT). Resistance associated with the Tyr181Cys mutation in HIV-1 RT has been a key roadblock in the discovery of nonnucleoside RT inhibitors (NNRTIs). It is the principal point mutation that arises from treatment of HIV-infected patients with nevirapine, the first-in-class drug still widely used, especially in developing countries. We report covalent inhibitors of Tyr181Cys RT (CRTIs) that can completely knock out activity of the resistant mutant and of the particularly challenging Lys103Asn/Tyr181Cys variant. Conclusive evidence for the covalent modification of Cys181 is provided from enzyme inhibition kinetics, mass spectrometry, protein crystallography, and antiviral activity in infected human T-cell assays. The CRTIs are also shown to be selective for Cys181 and have lower cytotoxicity than the approved NNRTI drugs efavirenz and rilpivirine.
PNAS 2017
doi: 10.1073/pnas.1711463114
Development of resistance remains a major challenge for drugs to treat HIV-1 infections, including those targeting the essential viral polymerase, HIV-1 reverse transcriptase (RT). Resistance associated with the Tyr181Cys mutation in HIV-1 RT has been a key roadblock in the discovery of nonnucleoside RT inhibitors (NNRTIs). It is the principal point mutation that arises from treatment of HIV-infected patients with nevirapine, the first-in-class drug still widely used, especially in developing countries. We report covalent inhibitors of Tyr181Cys RT (CRTIs) that can completely knock out activity of the resistant mutant and of the particularly challenging Lys103Asn/Tyr181Cys variant. Conclusive evidence for the covalent modification of Cys181 is provided from enzyme inhibition kinetics, mass spectrometry, protein crystallography, and antiviral activity in infected human T-cell assays. The CRTIs are also shown to be selective for Cys181 and have lower cytotoxicity than the approved NNRTI drugs efavirenz and rilpivirine.
Saturday, August 19, 2017
Road Map for the Structure-Based Design of Selective Covalent HCV NS3/4A Protease Inhibitors
Letitia Shunmugam, Pritika Ramharack, Mahmoud E. S. Soliman
The Protein Journal
https://link.springer.com/article/10.1007/s10930-017-9736-8
Over the last 2 decades, covalent inhibitors have gained much popularity and is living up to its reputation as a powerful tool in drug discovery. Covalent inhibitors possess many significant advantages including increased biochemical efficiency, prolonged duration and the ability to target shallow, solvent exposed substrate-binding domains. However, rapidly mounting concerns over the potential toxicity, highly reactive nature and general lack of selectivity have negatively impacted covalent inhibitor development. Recently, a great deal of emphasis by the pharmaceutical industry has been placed toward the development of novel approaches to alleviate the major challenges experienced through covalent inhibition. This has unexpectedly led to the emergence of “selective” covalent inhibitors. The purpose of this review is not only to provide an overview from literature but to introduce a technical guidance as to how to initiate a systematic “road map” for the design of selective covalent inhibitors which we believe may assist in the design and development of optimized potential selective covalent HCV NS3/4A viral protease inhibitors.
The Protein Journal
https://link.springer.com/article/10.1007/s10930-017-9736-8
Over the last 2 decades, covalent inhibitors have gained much popularity and is living up to its reputation as a powerful tool in drug discovery. Covalent inhibitors possess many significant advantages including increased biochemical efficiency, prolonged duration and the ability to target shallow, solvent exposed substrate-binding domains. However, rapidly mounting concerns over the potential toxicity, highly reactive nature and general lack of selectivity have negatively impacted covalent inhibitor development. Recently, a great deal of emphasis by the pharmaceutical industry has been placed toward the development of novel approaches to alleviate the major challenges experienced through covalent inhibition. This has unexpectedly led to the emergence of “selective” covalent inhibitors. The purpose of this review is not only to provide an overview from literature but to introduce a technical guidance as to how to initiate a systematic “road map” for the design of selective covalent inhibitors which we believe may assist in the design and development of optimized potential selective covalent HCV NS3/4A viral protease inhibitors.
Friday, August 18, 2017
Covalent Enzyme Inhibition through Fluorosulfate Modification of a Noncatalytic Serine Residue
Olugbeminiyi O. Fadeyi , Lise R. Hoth, Chulho Choi, Xidong Feng, Ariamala Gopalsamy, Erik C. Hett†∥, Robert E. Kyne Jr., Ralph P. Robinson, and Lyn H. Jones
ACS Chem. Biol., 2017, 12 (8), 2015–2020
DOI: 10.1021/acschembio.7b00403
Irreversible enzyme inhibitors and covalent chemical biology probes often utilize the reaction of a protein cysteine residue with an appropriately positioned electrophile (e.g., acrylamide) on the ligand template. However, cysteine residues are not always available for site-specific protein labeling, and therefore new approaches are needed to expand the toolkit of appropriate electrophiles (“warheads”) that target alternative amino acids. We previously described the rational targeting of tyrosine residues in the active site of a protein (the mRNA decapping scavenger enzyme, DcpS) using inhibitors armed with a sulfonyl fluoride electrophile. These inhibitors subsequently enabled the development of clickable probe technology to measure drug-target occupancy in live cells. Here we describe a fluorosulfate-containing inhibitor (aryl fluorosulfate probe (FS-p1)) with excellent chemical and metabolic stability that reacts selectively with a noncatalytic serine residue in the same active site of DcpS as confirmed by peptide mapping experiments. Our results suggest that noncatalytic serine targeting using fluorosulfate electrophilic warheads could be a suitable strategy for the development of covalent inhibitor drugs and chemical probes.
ACS Chem. Biol., 2017, 12 (8), 2015–2020
DOI: 10.1021/acschembio.7b00403
Irreversible enzyme inhibitors and covalent chemical biology probes often utilize the reaction of a protein cysteine residue with an appropriately positioned electrophile (e.g., acrylamide) on the ligand template. However, cysteine residues are not always available for site-specific protein labeling, and therefore new approaches are needed to expand the toolkit of appropriate electrophiles (“warheads”) that target alternative amino acids. We previously described the rational targeting of tyrosine residues in the active site of a protein (the mRNA decapping scavenger enzyme, DcpS) using inhibitors armed with a sulfonyl fluoride electrophile. These inhibitors subsequently enabled the development of clickable probe technology to measure drug-target occupancy in live cells. Here we describe a fluorosulfate-containing inhibitor (aryl fluorosulfate probe (FS-p1)) with excellent chemical and metabolic stability that reacts selectively with a noncatalytic serine residue in the same active site of DcpS as confirmed by peptide mapping experiments. Our results suggest that noncatalytic serine targeting using fluorosulfate electrophilic warheads could be a suitable strategy for the development of covalent inhibitor drugs and chemical probes.
Sunday, August 6, 2017
Development of Novel Peptide-based Michael Acceptors Targeting Rhodesain and Falcipain-2 for the Treatment of Neglected Tropical Diseases (NTDs)
Santo Preveti, Roberta Ettari, Sandro Cosconati, Giorgio Amendola, Khawla Chouchene, Annika Wagner, Ute A. Hellmich, Kathrin Ulrich, R. Luise Krauth-Siegel, Peter R. Wich, Ira Schmid, Tanja Schirmeister, Jiri Gut, Philip J. Rosenthal, Silvana Grasso, and Maria Zappalà
J. Med. Chem. 2017
DOI: 10.1021/acs.jmedchem.7b00405
This paper describes the development of a class of peptide-based inhibitors as novel antitrypanosomal and antimalarial agents. The inhibitors are based on a characteristic peptide-sequence for the inhibition of the cysteine proteases rhodesain of T. b. rhodesiense and falcipain-2 of P. falciparum. We exploited the reactivity of novel unsaturated electrophilic functions such as vinyl-sulfones, -ketones, -esters and –nitriles. The Michael acceptors inhibited both rhodesain and falcipain-2, at nanomolar and micromolar level, respectively. In particular, the vinyl ketone 3b has emerged as a potent rhodesain inhibitor (k2nd= 67•106 M-1 min-1), endowed with a picomolar binding affinity (Ki = 38 pM), coupled with a single-digit micromolar activity against T. b. brucei (EC50 = 2.97 µM), thus being considered as a novel lead compound for the discovery of novel effective antitrypanosomal agents.
J. Med. Chem. 2017
DOI: 10.1021/acs.jmedchem.7b00405
This paper describes the development of a class of peptide-based inhibitors as novel antitrypanosomal and antimalarial agents. The inhibitors are based on a characteristic peptide-sequence for the inhibition of the cysteine proteases rhodesain of T. b. rhodesiense and falcipain-2 of P. falciparum. We exploited the reactivity of novel unsaturated electrophilic functions such as vinyl-sulfones, -ketones, -esters and –nitriles. The Michael acceptors inhibited both rhodesain and falcipain-2, at nanomolar and micromolar level, respectively. In particular, the vinyl ketone 3b has emerged as a potent rhodesain inhibitor (k2nd= 67•106 M-1 min-1), endowed with a picomolar binding affinity (Ki = 38 pM), coupled with a single-digit micromolar activity against T. b. brucei (EC50 = 2.97 µM), thus being considered as a novel lead compound for the discovery of novel effective antitrypanosomal agents.
Saturday, August 5, 2017
Recent Developments in JAK3 Inhibition: Isoform Selectivity by Covalent Targeting of Cys909
Michael Forster, Matthias Gehringer, Stefan A. Laufer
doi: 10.1016/j.bmcl.2017.07.079
Janus kinases (JAKs) are a family of four cytosolic protein kinases with a high degree of structural similarity. Due to its very restricted role in immune regulation, JAK3 was promoted as an excellent target for immunosuppression for more than a decade, but clinical validation of this concept is still elusive. During the last years, speculation arose that kinase activity of JAK1, which cooperates with JAK3 in cytokine receptor signaling, may have a dominant role over the one of JAK3. Until recently, however, this issue could not be appropriately addressed due a lack of highly isoform-selective tool compounds. With the recent resurgence of covalent drugs, targeting of a specific cysteine that distinguishes JAK3 from the other JAK family members became an attractive design option. By applying this strategy, a set of JAK3 inhibitors with excellent selectivity against other JAK isoforms and the kinome was developed within the last three years and used to decipher JAK3-dependent signaling. The data obtained with these tool compounds demonstrates that selective JAK3 inhibition is sufficient to block downstream signaling. Since one of these inhibitors is currently under evaluation in phase II clinical studies against several inflammatory disorders, it will soon become apparent whether selective JAK3 inhibition translates into clinical efficacy.
doi: 10.1016/j.bmcl.2017.07.079
Janus kinases (JAKs) are a family of four cytosolic protein kinases with a high degree of structural similarity. Due to its very restricted role in immune regulation, JAK3 was promoted as an excellent target for immunosuppression for more than a decade, but clinical validation of this concept is still elusive. During the last years, speculation arose that kinase activity of JAK1, which cooperates with JAK3 in cytokine receptor signaling, may have a dominant role over the one of JAK3. Until recently, however, this issue could not be appropriately addressed due a lack of highly isoform-selective tool compounds. With the recent resurgence of covalent drugs, targeting of a specific cysteine that distinguishes JAK3 from the other JAK family members became an attractive design option. By applying this strategy, a set of JAK3 inhibitors with excellent selectivity against other JAK isoforms and the kinome was developed within the last three years and used to decipher JAK3-dependent signaling. The data obtained with these tool compounds demonstrates that selective JAK3 inhibition is sufficient to block downstream signaling. Since one of these inhibitors is currently under evaluation in phase II clinical studies against several inflammatory disorders, it will soon become apparent whether selective JAK3 inhibition translates into clinical efficacy.
Tuesday, August 1, 2017
Electrophilic Triterpenoid Enones: A Comparative Thiol-Trapping and Bioactivity Study
Danilo Del Prete, Orazio Taglialatela-Scafati, Alberto Minassi, Carmina Sirignano, Cristina Cruz, Maria L. Bellido, Eduardo Muñoz, and Giovanni Appendino
J. Nat. Prod., Article ASAP
DOI: 10.1021/acs.jnatprod.7b00271
Publication Date (Web): July 28, 2017
Bardoxolone methyl (1) is the quintessential member of triterpenoid cyanoacrylates, an emerging class of bioactive compounds capable of transient covalent binding to thiols. The mechanistic basis for this unusual “pulsed reactivity” profile and the mode of its biological translation are unknown. To provide clues on these issues, a series of Δ1-dehydrooleanolates bearing an electron-withdrawing group at C-2 (7a–m) were prepared from oleanolic acid (3a) and comparatively investigated in terms of reactivity with thiols and bioactivity against a series of electrophile-sensitive transcription factors (Nrf2, NF-κB, STAT3). The emerging picture suggests that the triterpenoid scaffold sharply decreases the reactivity of the enone system by steric encumbrance and that only strongly electrophilic and sterically undemanding substituents such as a cyanide or a carboxylate group can re-establish Michael reactivity, albeit in a transient way for the cyanide group. In general, a substantial dissection between the thiol-trapping ability and the modulation of biological end-points sensitive to thiol alkylation was observed, highlighting the role of shape complementarity for the activity of triterpenoid thia-Michael acceptors.
J. Nat. Prod., Article ASAP
DOI: 10.1021/acs.jnatprod.7b00271
Publication Date (Web): July 28, 2017
Bardoxolone methyl (1) is the quintessential member of triterpenoid cyanoacrylates, an emerging class of bioactive compounds capable of transient covalent binding to thiols. The mechanistic basis for this unusual “pulsed reactivity” profile and the mode of its biological translation are unknown. To provide clues on these issues, a series of Δ1-dehydrooleanolates bearing an electron-withdrawing group at C-2 (7a–m) were prepared from oleanolic acid (3a) and comparatively investigated in terms of reactivity with thiols and bioactivity against a series of electrophile-sensitive transcription factors (Nrf2, NF-κB, STAT3). The emerging picture suggests that the triterpenoid scaffold sharply decreases the reactivity of the enone system by steric encumbrance and that only strongly electrophilic and sterically undemanding substituents such as a cyanide or a carboxylate group can re-establish Michael reactivity, albeit in a transient way for the cyanide group. In general, a substantial dissection between the thiol-trapping ability and the modulation of biological end-points sensitive to thiol alkylation was observed, highlighting the role of shape complementarity for the activity of triterpenoid thia-Michael acceptors.
Saturday, July 29, 2017
Advances of small molecule targeting of kinases
Norbert Berndt, Rezaul M Karim, Ernst Schönbrunn
Current Opinion in Chemical Biology
Volume 39, August 2017, Pages 126–132
doi: 10.1016/j.cbpa.2017.06.015
Reversible protein phosphorylation regulates virtually all aspects of life in the cell. As a result, dysregulation of protein kinases, the enzymes responsible for transferring phosphate groups from ATP to proteins, are often the cause or consequence of many human diseases including cancer. Almost three dozen protein kinase inhibitors (PKIs) have been approved for clinical applications since 1995, the vast majority of them for the treatment of cancer. According to the NCI, there are more than 100 types of cancer. However, FDA-approved PKIs only target 14 of them. Importantly, of the more than 500 protein kinases encoded by the human genome, only 22 are targets for currently approved PKIs, suggesting that the reservoir of PKIs still has room to grow significantly. In this short review we will discuss the most recent advances, challenges, and alternatives to currently adopted strategies in this burgeoning field.
Current Opinion in Chemical Biology
Volume 39, August 2017, Pages 126–132
doi: 10.1016/j.cbpa.2017.06.015
Reversible protein phosphorylation regulates virtually all aspects of life in the cell. As a result, dysregulation of protein kinases, the enzymes responsible for transferring phosphate groups from ATP to proteins, are often the cause or consequence of many human diseases including cancer. Almost three dozen protein kinase inhibitors (PKIs) have been approved for clinical applications since 1995, the vast majority of them for the treatment of cancer. According to the NCI, there are more than 100 types of cancer. However, FDA-approved PKIs only target 14 of them. Importantly, of the more than 500 protein kinases encoded by the human genome, only 22 are targets for currently approved PKIs, suggesting that the reservoir of PKIs still has room to grow significantly. In this short review we will discuss the most recent advances, challenges, and alternatives to currently adopted strategies in this burgeoning field.
Friday, July 21, 2017
Covalent Enzyme Inhibition through Fluorosulfate Modification of a Noncatalytic Serine Residue
Olugbeminiyi O. Fadeyi, Lise R. Hoth, Chulho Choi, Xidong Feng, Ariamala Gopalsamy, Erik C. Hett, Robert E. Kyne Jr., Ralph P. Robinson, and Lyn H. Jones
Medicine Design, Pfizer Inc., 610 Main Street, Cambridge, Massachusetts 02139, United States
Medicine Design, Pfizer Inc., Eastern Point Road, Groton, Connecticut 06340, United States
ACS Chem. Biol., Article ASAP
DOI: 10.1021/acschembio.7b00403
Irreversible enzyme inhibitors and covalent chemical biology probes often utilize the reaction of a protein cysteine residue with an appropriately positioned electrophile (e.g., acrylamide) on the ligand template. However, cysteine residues are not always available for site-specific protein labeling, and therefore new approaches are needed to expand the toolkit of appropriate electrophiles (“warheads”) that target alternative amino acids. We previously described the rational targeting of tyrosine residues in the active site of a protein (the mRNA decapping scavenger enzyme, DcpS) using inhibitors armed with a sulfonyl fluoride electrophile. These inhibitors subsequently enabled the development of clickable probe technology to measure drug-target occupancy in live cells. Here we describe a fluorosulfate-containing inhibitor (aryl fluorosulfate probe (FS-p1)) with excellent chemical and metabolic stability that reacts selectively with a noncatalytic serine residue in the same active site of DcpS as confirmed by peptide mapping experiments. Our results suggest that noncatalytic serine targeting using fluorosulfate electrophilic warheads could be a suitable strategy for the development of covalent inhibitor drugs and chemical probes.
Medicine Design, Pfizer Inc., 610 Main Street, Cambridge, Massachusetts 02139, United States
Medicine Design, Pfizer Inc., Eastern Point Road, Groton, Connecticut 06340, United States
ACS Chem. Biol., Article ASAP
DOI: 10.1021/acschembio.7b00403
Irreversible enzyme inhibitors and covalent chemical biology probes often utilize the reaction of a protein cysteine residue with an appropriately positioned electrophile (e.g., acrylamide) on the ligand template. However, cysteine residues are not always available for site-specific protein labeling, and therefore new approaches are needed to expand the toolkit of appropriate electrophiles (“warheads”) that target alternative amino acids. We previously described the rational targeting of tyrosine residues in the active site of a protein (the mRNA decapping scavenger enzyme, DcpS) using inhibitors armed with a sulfonyl fluoride electrophile. These inhibitors subsequently enabled the development of clickable probe technology to measure drug-target occupancy in live cells. Here we describe a fluorosulfate-containing inhibitor (aryl fluorosulfate probe (FS-p1)) with excellent chemical and metabolic stability that reacts selectively with a noncatalytic serine residue in the same active site of DcpS as confirmed by peptide mapping experiments. Our results suggest that noncatalytic serine targeting using fluorosulfate electrophilic warheads could be a suitable strategy for the development of covalent inhibitor drugs and chemical probes.
Thursday, July 20, 2017
Discovery of a covalent kinase inhibitor from a DNA encoded small-molecule library X protein library selection
Discovery of a Covalent Kinase Inhibitor from a DNA-Encoded Small-Molecule Library × Protein Library Selection
Alix I. Chan, Lynn M. McGregor, Tara Jain, and David R. Liu
The Broad Institute of Harvard and MIT, Howard Hughes Medical Institute, and the Department of Chemistry and Chemical Biology, Harvard University, 75 Ames Street, Cambridge, Massachusetts 02142, United States
J. Am. Chem. Soc., Article ASAP
DOI: 10.1021/jacs.7b04880
We previously reported interaction determination using unpurified proteins (IDUP), a method to selectively amplify DNA sequences encoding ligand:target pairs from a mixture of DNA-linked small molecules and unpurified protein targets in cell lysates. In this study, we applied IDUP to libraries of DNA-encoded bioactive compounds and DNA-tagged human kinases to identify ligand:protein binding partners out of 32 096 possible combinations in a single solution-phase library × library experiment. The results recapitulated known small molecule:protein interactions and also revealed that ethacrynic acid is a novel ligand and inhibitor of MAP2K6 kinase. Ethacrynic acid inhibits MAP2K6 in part through alkylation of a nonconserved cysteine residue. This work validates the ability of IDUP to discover ligands for proteins of biomedical relevance.
Alix I. Chan, Lynn M. McGregor, Tara Jain, and David R. Liu
The Broad Institute of Harvard and MIT, Howard Hughes Medical Institute, and the Department of Chemistry and Chemical Biology, Harvard University, 75 Ames Street, Cambridge, Massachusetts 02142, United States
J. Am. Chem. Soc., Article ASAP
DOI: 10.1021/jacs.7b04880
We previously reported interaction determination using unpurified proteins (IDUP), a method to selectively amplify DNA sequences encoding ligand:target pairs from a mixture of DNA-linked small molecules and unpurified protein targets in cell lysates. In this study, we applied IDUP to libraries of DNA-encoded bioactive compounds and DNA-tagged human kinases to identify ligand:protein binding partners out of 32 096 possible combinations in a single solution-phase library × library experiment. The results recapitulated known small molecule:protein interactions and also revealed that ethacrynic acid is a novel ligand and inhibitor of MAP2K6 kinase. Ethacrynic acid inhibits MAP2K6 in part through alkylation of a nonconserved cysteine residue. This work validates the ability of IDUP to discover ligands for proteins of biomedical relevance.
Sunday, July 16, 2017
1,6-Cyclophellitol Cyclosulfates: A New Class of Irreversible Glycosidase Inhibitor
Marta Artola , Liang Wu, Maria J. Ferraz, Chi-Lin Kuo§, Lluís Raich∥, Imogen Z. Bree‡, Wendy A. Offen, Jeroen D. C. Codée , Gijsbert A. van der Marel, Carme Rovira, Johannes M. F. G. Aerts, Gideon J. Davies , and Herman S. Overkleeft
ACS Cent. Sci., Article ASAP
DOI: 10.1021/acscentsci.7b00214
The essential biological roles played by glycosidases, coupled to the diverse therapeutic benefits of pharmacologically targeting these enzymes, provide considerable motivation for the development of new inhibitor classes. Cyclophellitol epoxides and aziridines are recently established covalent glycosidase inactivators. Inspired by the application of cyclic sulfates as electrophilic equivalents of epoxides in organic synthesis, we sought to test whether cyclophellitol cyclosulfates would similarly act as irreversible glycosidase inhibitors. Here we present the synthesis, conformational analysis, and application of novel 1,6-cyclophellitol cyclosulfates. We show that 1,6-epi-cyclophellitol cyclosulfate (α-cyclosulfate) is a rapidly reacting α-glucosidase inhibitor whose 4C1 chair conformation matches that adopted by α-glucosidase Michaelis complexes. The 1,6-cyclophellitol cyclosulfate (β-cyclosulfate) reacts more slowly, likely reflecting its conformational restrictions. Selective glycosidase inhibitors are invaluable as mechanistic probes and therapeutic agents, and we propose cyclophellitol cyclosulfates as a valuable new class of carbohydrate mimetics for application in these directions.
ACS Cent. Sci., Article ASAP
DOI: 10.1021/acscentsci.7b00214
The essential biological roles played by glycosidases, coupled to the diverse therapeutic benefits of pharmacologically targeting these enzymes, provide considerable motivation for the development of new inhibitor classes. Cyclophellitol epoxides and aziridines are recently established covalent glycosidase inactivators. Inspired by the application of cyclic sulfates as electrophilic equivalents of epoxides in organic synthesis, we sought to test whether cyclophellitol cyclosulfates would similarly act as irreversible glycosidase inhibitors. Here we present the synthesis, conformational analysis, and application of novel 1,6-cyclophellitol cyclosulfates. We show that 1,6-epi-cyclophellitol cyclosulfate (α-cyclosulfate) is a rapidly reacting α-glucosidase inhibitor whose 4C1 chair conformation matches that adopted by α-glucosidase Michaelis complexes. The 1,6-cyclophellitol cyclosulfate (β-cyclosulfate) reacts more slowly, likely reflecting its conformational restrictions. Selective glycosidase inhibitors are invaluable as mechanistic probes and therapeutic agents, and we propose cyclophellitol cyclosulfates as a valuable new class of carbohydrate mimetics for application in these directions.
Tuesday, July 11, 2017
Discovery of Heteroaromatic Sulfones As A New Class of Biologically Compatible Thiol-Selective Reagents
Xiaofei Chen, Hanzhi Wu, Chung-Min Park, Thomas H. Poole, Gizem Keceli, Nelmi O. Devarie Baez, Allen W. Tsang, W. Todd Lowther, Leslie B. Poole, S. Bruce King, Ming Xian, and Cristina M. Furdui
ACS Chem. Biol., 2017
DOI: 10.1021/acschembio.7b00444
The selective reaction of chemical reagents with reduced protein thiols is critical to biological research. This reaction is utilized to prevent crosslinking of cysteine-containing peptides in common proteomics workflows and is applied widely in discovery and targeted redox investigations of the mechanisms underlying physiological and pathological processes. However, known and commonly used thiol blocking reagents like iodoacetamide, N-ethylmaleimide and others were found to cross-react with oxidized protein sulfenic acids (-SOH) introducing significant errors in studies employing these reagents. We have investigated and are reporting here a new heteroaromatic alkylsulfone, 4-(5-Methanesulfonyl-[1,2,3,4]tetrazol-1-yl)-phenol (MSTP), as selective and highly reactive -SH blocking reagent compatible with biological applications.
ACS Chem. Biol., 2017
DOI: 10.1021/acschembio.7b00444
The selective reaction of chemical reagents with reduced protein thiols is critical to biological research. This reaction is utilized to prevent crosslinking of cysteine-containing peptides in common proteomics workflows and is applied widely in discovery and targeted redox investigations of the mechanisms underlying physiological and pathological processes. However, known and commonly used thiol blocking reagents like iodoacetamide, N-ethylmaleimide and others were found to cross-react with oxidized protein sulfenic acids (-SOH) introducing significant errors in studies employing these reagents. We have investigated and are reporting here a new heteroaromatic alkylsulfone, 4-(5-Methanesulfonyl-[1,2,3,4]tetrazol-1-yl)-phenol (MSTP), as selective and highly reactive -SH blocking reagent compatible with biological applications.
Monday, July 3, 2017
Drug-Target Kinetics in Drug Discovery
Peter J Tonge
ACS Chem. Neurosci., Just Accepted Manuscript
DOI: 10.1021/acschemneuro.7b00185
The development of therapies for the treatment of neurological cancer faces a number of major challenges including the synthesis of small molecule agents that can penetrate the blood brain barrier (BBB). Given the likelihood that in many cases drug exposure will be lower in the CNS than in systemic circulation, it follows that strategies should be employed that can sustain target engagement at low drug concentration. Time dependent target occupancy is a function of both the drug and target concentration as well as the thermodynamic and kinetic parameters that describe the binding reaction coordinate, and sustained target occupancy can be achieved through structural modifications that increase target (re)binding and/or that decrease the rate of drug dissociation. The discovery and deployment of compounds with optimized kinetic effects requires information on the structure-kinetic relationships that modulate the kinetics of binding, and the molecular factors that control the translation of drug-target kinetics to time-dependent drug activity in the disease state. This review first introduces the potential benefits of drug-target kinetics, such as the ability to delineate both thermodynamic and kinetic selectivity, and then describes factors, such as target vulnerability, that impact the utility of kinetic selectivity. The review concludes with a description of a mechanistic PK/PD model that integrates drug-target kinetics into predictions of drug activity.
ACS Chem. Neurosci., Just Accepted Manuscript
DOI: 10.1021/acschemneuro.7b00185
The development of therapies for the treatment of neurological cancer faces a number of major challenges including the synthesis of small molecule agents that can penetrate the blood brain barrier (BBB). Given the likelihood that in many cases drug exposure will be lower in the CNS than in systemic circulation, it follows that strategies should be employed that can sustain target engagement at low drug concentration. Time dependent target occupancy is a function of both the drug and target concentration as well as the thermodynamic and kinetic parameters that describe the binding reaction coordinate, and sustained target occupancy can be achieved through structural modifications that increase target (re)binding and/or that decrease the rate of drug dissociation. The discovery and deployment of compounds with optimized kinetic effects requires information on the structure-kinetic relationships that modulate the kinetics of binding, and the molecular factors that control the translation of drug-target kinetics to time-dependent drug activity in the disease state. This review first introduces the potential benefits of drug-target kinetics, such as the ability to delineate both thermodynamic and kinetic selectivity, and then describes factors, such as target vulnerability, that impact the utility of kinetic selectivity. The review concludes with a description of a mechanistic PK/PD model that integrates drug-target kinetics into predictions of drug activity.
Tuesday, June 27, 2017
Privileged Electrophile Sensors: A Resource for Covalent Drug Development
Marcus John Curtis Long, Yimon Aye
Cell Chemical Biology
10.1016/j.chembiol.2017.05.023
This Perspective delineates how redox signaling affects the activity of specific enzyme isoforms and how this property may be harnessed for rational drug design. Covalent drugs have resurged in recent years and several reports have extolled the general virtues of developing irreversible inhibitors. Indeed, many modern pharmaceuticals contain electrophilic appendages. Several invoke a warhead that hijacks active-site nucleophiles whereas others take advantage of spectator nucleophilic side chains that do not participate in enzymatic chemistry, but are poised to bind/react with electrophiles. The latest data suggest that innate electrophile sensing—which enables rapid reaction with an endogenous signaling electrophile—is a quintessential resource for the development of covalent drugs. For instance, based on recent work documenting isoform-specific electrophile sensing, isozyme non-specific drugs may be converted to isozyme-specific analogs by hijacking privileged first-responder electrophile-sensing cysteines. Because this approach targets functionally relevant cysteines, we can simultaneously harness previously untapped moonlighting roles of enzymes linked to redox sensing
Cell Chemical Biology
10.1016/j.chembiol.2017.05.023
This Perspective delineates how redox signaling affects the activity of specific enzyme isoforms and how this property may be harnessed for rational drug design. Covalent drugs have resurged in recent years and several reports have extolled the general virtues of developing irreversible inhibitors. Indeed, many modern pharmaceuticals contain electrophilic appendages. Several invoke a warhead that hijacks active-site nucleophiles whereas others take advantage of spectator nucleophilic side chains that do not participate in enzymatic chemistry, but are poised to bind/react with electrophiles. The latest data suggest that innate electrophile sensing—which enables rapid reaction with an endogenous signaling electrophile—is a quintessential resource for the development of covalent drugs. For instance, based on recent work documenting isoform-specific electrophile sensing, isozyme non-specific drugs may be converted to isozyme-specific analogs by hijacking privileged first-responder electrophile-sensing cysteines. Because this approach targets functionally relevant cysteines, we can simultaneously harness previously untapped moonlighting roles of enzymes linked to redox sensing
Residue-Specific Peptide Modification: A Chemist’s Guide
Justine N. deGruyter, Lara R. Malins, and Phil S. Baran
Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037
DOI: 10.1021/acs.biochem.7b00536
Advances in bioconjugation and native protein modification are appearing at a blistering pace, making it increasingly time consuming for practitioners to identify the best chemical method to modify a specific amino acid residue in a complex setting. The purpose of this perspective is to provide an informative, graphically-rich manual highlighting significant advances in the field over the past decade. This guide will help triage candidate methods for peptide alteration, and will serve as a starting point for those seeking to solve longstanding challenges.
Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037
DOI: 10.1021/acs.biochem.7b00536
Advances in bioconjugation and native protein modification are appearing at a blistering pace, making it increasingly time consuming for practitioners to identify the best chemical method to modify a specific amino acid residue in a complex setting. The purpose of this perspective is to provide an informative, graphically-rich manual highlighting significant advances in the field over the past decade. This guide will help triage candidate methods for peptide alteration, and will serve as a starting point for those seeking to solve longstanding challenges.
Wednesday, June 21, 2017
2-Chloropropionamide as a low-reactivity electrophile for irreversible small-molecule probe identification
Dharmaraja Allimuthu and Drew J. Adams
ACS Chem. Biol., 2017, Just Accepted Manuscript
DOI: 10.1021/acschembio.7b00424
ACS Chem. Biol., 2017, Just Accepted Manuscript
DOI: 10.1021/acschembio.7b00424
Abstract
Resurgent interest in covalent target engagement in drug discovery has demonstrated that small molecules containing weakly reactive electrophiles can be safe and effective therapies. Several recently FDA-approved drugs feature an acrylamide functionality to selectively engage cysteine side chains of kinases (Ibrutinib, Afatinib, and Neratinib). Additional electrophilic functionalities whose reactivity is compatible with highly selective target engagement and in vivo application could open new avenues in covalent small molecule discovery. Here we report the synthesis and evaluation of a library of small molecules containing the 2-chloropropionamide functionality, which we demonstrate is less reactive than typical acrylamide electrophiles. Although many library members do not appear to label proteins in cells, we identified S-CW3554 as selectively labeling protein disulfide isomerase and inhibiting its enzymatic activity. Subsequent profiling of the library against five diverse cancer cell lines showed unique cytotoxicity for S-CW3554 in cells derived from multiple myeloma, a cancer recently reported to be sensitive to PDI inhibition. Our novel PDI inhibitor highlights the potential of 2-chloropropionamides as weak and stereochemically-tunable electrophiles for covalent drug discovery.Tuesday, June 20, 2017
Farnesyltransferase-Mediated Delivery of a Covalent Inhibitor Overcomes Alternative Prenylation to Mislocalize K-Ras
Chris J. Novotny, Gregory L. Hamilton, Frank McCormick, and Kevan M. Shokat
† Howard Hughes Medical Institute and Department of Cellular and Molecular Pharmacology, University of California San Francisco, San Francisco, California 94158, United States
‡ NCI RAS Initiative, Cancer Research Technology Program, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research, Inc., Frederick, Maryland 21701, United States
§ Diller Family Comprehensive Cancer Center, University of California, San Francisco, California 94158, United States
ACS Chem. Biol., Article ASAP
DOI: 10.1021/acschembio.7b00374
Mutationally activated Ras is one of the most common oncogenic drivers found across all malignancies, and its selective inhibition has long been a goal in both pharma and academia. One of the oldest and most validated methods to inhibit overactive Ras signaling is by interfering with its post-translational processing and subsequent cellular localization. Previous attempts to target Ras processing led to the development of farnesyltransferase inhibitors, which can inhibit H-Ras localization but not K-Ras due to its ability to bypass farnesyltransterase inhibition through alternative prenylation by geranylgeranyltransferase. Here, we present the creation of a neo-substrate for farnesyltransferase that prevents the alternative prenlation by geranylgeranyltransferase and mislocalizes oncogenic K-Ras in cells.
† Howard Hughes Medical Institute and Department of Cellular and Molecular Pharmacology, University of California San Francisco, San Francisco, California 94158, United States
‡ NCI RAS Initiative, Cancer Research Technology Program, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research, Inc., Frederick, Maryland 21701, United States
§ Diller Family Comprehensive Cancer Center, University of California, San Francisco, California 94158, United States
ACS Chem. Biol., Article ASAP
DOI: 10.1021/acschembio.7b00374
Mutationally activated Ras is one of the most common oncogenic drivers found across all malignancies, and its selective inhibition has long been a goal in both pharma and academia. One of the oldest and most validated methods to inhibit overactive Ras signaling is by interfering with its post-translational processing and subsequent cellular localization. Previous attempts to target Ras processing led to the development of farnesyltransferase inhibitors, which can inhibit H-Ras localization but not K-Ras due to its ability to bypass farnesyltransterase inhibition through alternative prenylation by geranylgeranyltransferase. Here, we present the creation of a neo-substrate for farnesyltransferase that prevents the alternative prenlation by geranylgeranyltransferase and mislocalizes oncogenic K-Ras in cells.
Saturday, June 17, 2017
Expanding the Scope of Electrophiles Capable of Targeting K-Ras Oncogenes
Lynn M. McGregor, Meredith L. Jenkins, Caitlin Kerwin, John E. Burke, and Kevan M. Shokat
Biochemistry, Article ASAP
DOI: 10.1021/acs.biochem.7b00271
There is growing interest in reversible and irreversible covalent inhibitors that target noncatalytic amino acids in target proteins. With a goal of targeting oncogenic K-Ras variants (e.g., G12D) by expanding the types of amino acids that can be targeted by covalent inhibitors, we survey a set of electrophiles for their ability to label carboxylates. We functionalized an optimized ligand for the K-Ras switch II pocket with a set of electrophiles previously reported to react with carboxylates and characterized the ability of these compounds to react with model nucleophiles and oncogenic K-Ras proteins. Here, we report that aziridines and stabilized diazo groups preferentially react with free carboxylates over thiols. Although we did not identify a warhead that potently labels K-Ras G12D, we were able to study the interactions of many electrophiles with K-Ras, as most of the electrophiles rapidly label K-Ras G12C. We characterized the resulting complexes by crystallography, hydrogen/deuterium exchange, and differential scanning fluorimetry. Our results both demonstrate the ability of a noncatalytic cysteine to react with a diverse set of electrophiles and emphasize the importance of proper spatial arrangements between a covalent inhibitor and its intended nucleophile. We hope that these results can expand the range of electrophiles and nucleophiles of use in covalent protein modulation.
Biochemistry, Article ASAP
DOI: 10.1021/acs.biochem.7b00271
There is growing interest in reversible and irreversible covalent inhibitors that target noncatalytic amino acids in target proteins. With a goal of targeting oncogenic K-Ras variants (e.g., G12D) by expanding the types of amino acids that can be targeted by covalent inhibitors, we survey a set of electrophiles for their ability to label carboxylates. We functionalized an optimized ligand for the K-Ras switch II pocket with a set of electrophiles previously reported to react with carboxylates and characterized the ability of these compounds to react with model nucleophiles and oncogenic K-Ras proteins. Here, we report that aziridines and stabilized diazo groups preferentially react with free carboxylates over thiols. Although we did not identify a warhead that potently labels K-Ras G12D, we were able to study the interactions of many electrophiles with K-Ras, as most of the electrophiles rapidly label K-Ras G12C. We characterized the resulting complexes by crystallography, hydrogen/deuterium exchange, and differential scanning fluorimetry. Our results both demonstrate the ability of a noncatalytic cysteine to react with a diverse set of electrophiles and emphasize the importance of proper spatial arrangements between a covalent inhibitor and its intended nucleophile. We hope that these results can expand the range of electrophiles and nucleophiles of use in covalent protein modulation.
Wednesday, June 14, 2017
Structure-activity relationship investigation for benzonaphthyridinone derivatives as novel potent Bruton's tyrosine kinase (BTK) irreversible inhibitors
Beilei Wanga, Yuanxin Denga, Yongfei Chena, Kailin Yua, Aoli Wanga, Qianmao Lianga, Wei Wanga, Cheng Chena, Hong Wua, Chen Hua, Weili Miaod, Wooyoung Hure, Wenchao Wanga
doi: 10.1016/j.ejmech.2017.06.016
doi: 10.1016/j.ejmech.2017.06.016
Abstract
Through a structure-based drug design approach, a tricyclic benzonaphthyridinone pharmacophore was used as a starting point for carrying out detailed medicinal structure-activity relationhip (SAR) studies geared toward characterization of a panel of proposed BTK inhibitors, including 6 (QL-X-138), 7 (BMX-IN-1) and 8 (QL-47). These studies led to the discovery of the novel potent irreversible BTK inhibitor, compound 18 (CHMFL-BTK-11). Kinetic analysis of compound 18 revealed an irreversible binding efficacy (kinact/Ki) of 0.01 μM−1s−1. Compound 18 potently inhibited BTK kinase Y223 auto-phosphorylation (EC50 < 100 nM), arrested cell cycle in G0/G1 phase, and induced apoptosis in Ramos, MOLM13 and Pfeiffer cells. We believe these features would make 18 a good pharmacological tool for studying BTK-related pathologies.
Tuesday, June 13, 2017
Covalent inhibitors design and discovery
Stephane De Cesco, Jerry Kurian, Caroline Dufresne, Anthony Mittermaier, Nicolas Moitessier
doi: 10.1016/j.ejmech.2017.06.019
doi: 10.1016/j.ejmech.2017.06.019
Abstract
In the history of therapeutics, covalent drugs occupy a very distinct category. While representing a significant fraction of the drugs on the market, very few have been deliberately designed to interact covalently with their biological target. In this review, the prevalence of covalent drugs will first be briefly covered, followed by an introduction to their mechanisms of action and more detailed discussions of their discovery and the development of safe and efficient covalent enzyme inhibitors. All stages of a drug discovery program will be covered, from target considerations to lead optimization, strategies to tune reactivity and computational methods. The goal of this article is to provide an overview of the field and to outline good practices that are needed for the proper assessment and development of covalent inhibition as well as good understanding of the potential and limitations of current computational methods for the design of covalent drugs.Saturday, June 10, 2017
Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10-2474
Subscribe to:
Comments (Atom)
-
Design, synthesis and biological evaluation of the activity-based probes for FGFR covalent inhibitorDandan Zhu, Zijian Zheng, Huixin Huang, Xiaojuan Chen, Shuhong Zhang, Zhuchu Chen, Ting Liu, Guangyu Xu, Ying Fu, Yongheng Chen, European Jo...
-
DOI Ansgar Oberheide, Maxime van den Oetelaar, Jakob Scheele, Jan Borggräfe, Semmy Engelen, Michael Sattler, Christian Ottmann, ...
-
Nafizul Haque Kazi, Nikolas Klink, Kai Gallant, Gian-Marvin Kipka & Malte Gersch Nat Struct Mol Biol , 2025 https://doi.org/10.1038/s415...
